
Abstract
Visual impairment is commonly reported as a consequence of heavy prenatal ethanol exposure in humans. Children generally display characteristic cranio-facial dysmorphology and represent typical severe cases of foetal alcohol syndrome. Binge-like rodent model systems have concluded that third trimester equivalent ethanol exposure results in widespread apoptosis in the visual system from the retina to the visual cortex. Neither clinical nor animal studies address the consequences of more moderate prenatal ethanol exposure on the visual system. The current study uses a naturalistic and voluntary consumption approach in non-human primates (Chlorocebus sabeus) in order to more closely model prenatal ethanol consumption patterns in humans. Pregnant vervet monkeys voluntarily drank on average 2.418 ± 0.296 g etoh/kg/day four times a week during the third trimester. Using unbiased stereology, we estimated the neuronal and glial population of the parvocellular (P) and magnocellular (M) layers of the lateral geniculate nucleus (LGN) following foetal alcohol exposure (FAE) in infant subjects. Layer volume and total number of neurons and glia in the LGN of the FAE subjects were not significantly different from age-matched control subjects. The M neuronal soma size of FAE subjects, however, was significantly reduced to resemble the size of the P-neurons. These results suggest that alterations at the level of morphology and anatomy of the M-neurons may lead to behavioural deficits associated with the integrity of the dorsal visual pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Foetal alcohol syndrome (FAS), a consequence of heavy maternal drinking during organogenesis, is readily recognizable in the form of distinct facial dysmorphologies such as microcephaly, indistinct philtrum and low nasal bridge (Streissguth et al. 1994). These dysmorphologies are typically a consequence of early and high levels of prenatal alcohol exposure. In addition, there are developmental impairments associated with more moderate prenatal ethanol exposure, the so-called non-dysmorphic FAS (Streissguth et al. 1994; Riley et al. 2004; Riley and McGee 2005) and can be grouped under the umbrella term foetal alcohol spectrum disorders (FASD). Foetal alcohol exposure is a leading cause of preventable developmental disorders (Mattson et al. 2001) with up to 10% of pregnant women reporting consuming moderate amounts of ethanol during pregnancy (Sidhu and Floyd 2002; Denny et al. 2009) and between 1 and 4% of live births being diagnosed as FAS (Sampson et al. 1997; Floyd and Sidhu 2004; Colvin et al. 2007; Kristjanson et al. 2007).
It has been estimated that up to 90% of FAS children have ocular manifestations. (Stromland 1985; Stromland and Pinazo-Duran 2002). Although no single visual abnormality can be considered as a defining characteristic of FAS, there are a number of manifestations of prenatal ethanol exposure in the visual system. Increased tortuosity of the retinal vessels (49%), suboptimal vision ranging from severe myopia to moderate hyperopia (65%), strabismus (43%) and hypoplasia of the optic nerve (48%) are commonly reported in FAS children (Stromland 1985; Cook et al. 1987; Stromland and Pinazo-Duran 2002). A recent clinical study that examined children aged between 4 and 9, concluded that the risks of ocular deficits are limited to children who were exposed to high levels of prenatal ethanol and developed facial dysmorphologies (Flanigan et al. 2008).
Rodent models of FAS suggest widespread apoptosis throughout the visual system following binge-like exposure (Tenkova et al. 2003). In clinical cases, magnetic resonance imaging (MRI) studies have found an overall reduction in brain size (Spadoni et al. 2007) of FASD individuals. There is no clear reduction in size of the thalamus and the occipital cortex (Archibald et al. 2001), but positron emission tomography (PET) has revealed reduced metabolic rates in the thalamus (Clark et al. 2000) which could indicate a selective toxic alcohol effect that may be either region- or cell-specific. Neither clinical nor animal model systems address the effects of the more common moderate prenatal ethanol exposure on the visual system.
The use of non-human primates provides an excellent translational model for our understanding of developmental processes (Riley and McGee 2005) given the developmental similarities between monkeys and humans (Garey and de Courten 1983). Few studies have taken advantage of the non-human primate as a model of FAE, especially concerning the effects of moderate prenatal alcohol exposure on the visual system. Weekly binge-like exposure throughout pregnancy in the non-human primate is reported to result in cell loss in the retinal ganglion layer in a subset of subjects (Clarren et al. 1990). In both clinical and animal model systems, the effects of prenatal ethanol exposure on the LGN have not been specifically examined. This current study takes advantage of moderate and naturalistic drinking patterns in the vervet monkey (Chlorocebus sabeus) to model the type of exposure frequently reported in human epidemiological investigations (Tsai and Floyd 2004). We tested the hypothesis that vervet monkeys exposed to ethanol during the last half of pregnancy would have an altered neuronal population in the lateral geniculate nucleus at birth.
Materials and methods
Maternal ethanol exposure
Healthy adult female vervet monkeys (Chlorocebus sabeus) were screened for voluntary ethanol consumption. Females that reliably drank at least 2 g ethanol/kg body weight in a 4-h scheduled period were selected for the study according to previously established protocols (Burke et al. 2009b). Females were monitored behaviourally and physically for evidence that pregnancy was initiated and then examined every month for timing of gestation.
At about embryonic day 89 of the normal 165-day gestational period, pregnant females were given access to a maximum of 3 g ethanol/kg body weight (or an isocaloric sucrose-control mixture) at 9 a.m. on 4 days of the week (Monday, Tuesday, Thursday, Friday) according to Burke et al. (2009b). Ethanol was prepared as a 10% v/v solution and presented in a calibrated drinking bottle with tap water concurrently available. The ethanol bottle was monitored every hour, and remained available for a 4-h period and the quantity consumed was recorded. After parturition, the consumption level for each mother was calculated on the basis of the total volume of ethanol consumed and expressed as g/kg body weight/drinking day, as well as total exposure (g/kg) over the gestational period. Blood (1 ml, saphenous vein) was drawn without anaesthesia at the end of the drinking period during weeks 2, 4, 6, and 8 for the measurement of blood ethanol level (alcohol dehydrogenase method, Sigma). Maternal blood ethanol concentrations are similar to that found in humans after consuming 3–5 standard drinks (Table 1). Animals were housed socially in the laboratories of the Behavioural Sciences Foundation (BSF), St Kitts in enriched environments. The animals were fed Harlan Teklad high-protein primate chow (5% body weight/day) and fresh local fruit, with water available ad libitum. The experimental protocol was reviewed and approved by the McGill University Animal Care and Use Committee and the Institutional Review Board of BSF.
Ethanol-exposed offspring
The infant monkey is estimated to have the same neurodevelopmental equivalency to a late term/neonatal human (Clancy et al. 2001). In the current study, 5 infant alcohol-exposed animals (4 m, 1f) and 5 infant sucrose-control (5 m) animals were chosen for neuroanatomical evaluation (Table 1). None of the offspring displayed craniofacial dysmorphology and were group-housed with their mothers until they were sacrificed. These subjects are part of an on-going research programme investigating the behavioural and neuroanatomical effects of prenatal ethanol exposure in the non-human primate.
Each animal was sedated with ketamine (10 mg/kg, i.m.), deeply anaesthetised with sodium pentobarbital (25 mg/kg, i.v.) and perfused transcardially with phosphate buffer saline (PBS), followed by 4% paraformaldehyde. The brains were then extracted, stereotaxically blocked into 1-cm slabs, cryoprotected in buffered sucrose, and frozen at −80°C (Burke et al. 2009c). Six parallel series of coronal sections (50 μm) were obtained for each animal with one series stained with cresyl-violet and the other series were placed in antigen preserve (50% ethylene glycol, phosphate buffer solution, and 1% polyvinyl pyrrolodone) and stored at −20°C (Burke et al. 2009d).
Stereology
Unbiased estimation of neuron and glia numbers along with layer volume was performed by using the optical fractionator method (West et al. 1991). Topography and superimposed counting frames (disectors) were generated through Stereologer software under 2.5× (topography) and 100× objectives (N.A. = 1.3). The lateral geniculate nucleus is readily identifiable as a six-layer structure dorsal to the hippocampal formation (Paxinos et al. 1999; Fig. 3). For the stereological parameters in this study, every 12th section was selected with a random starting point within 1 mm of the anterior tip that yielded a systematic-uniform set sample of 9–10 sections. The LGN was sampled into its subdivisions: M (layers 1–2) and P (layers 3–6). Total reference volume (V) was achieved using the Cavalieri point counting method. A standard x–y grid size of 100 μm was used to calculate the volume of M and P regions (see Table 2 for the stereological parameters).
The total estimation of cell numbers (N) was calculated by the following equation:
where ssf is the section sampling fraction, asf is the area sampling fraction, tsf is the thickness-sampling fraction (where the measured thickness of the tissue is divided by the disector height), and ∑Q − is the total number of cells counted within a disector. For this study, a neuron was defined as having visible and centrally located nucleoli and clearly defined cytoplasm (Boire et al. 2002; Joelving et al. 2006; Burke et al. 2009a, b). The x–y step was set at 350 and 750 μm for M and P regions, respectively. Coefficients of error (CE) were calculated for mean number of neurons and glia (∑Q −), total number of disectors (∑F), and total number of cells (N) to assess the reliability of measurements (Gundersen and Jensen 1987; West and Gundersen 1990). As the CE represents intrinsic methodological uncertainty, its contribution to observed variation (CV) should be less than its contribution to biological variation (BCV). The ratio BCV2/CV2, where BCV2 = CV2—mean CE2 and CV2 = BCV2 + CE2, was used to determine the precision of the estimates (West et al. 1991). A ratio BCV2/CV2 of more than 0.5 indicates acceptable stereological precision (Joelving et al. 2006).
Neuronal soma volume was determined using the isotropic-uniform-random (IUR) nuclear rotator parameter of the Stereologer system. Neurons that were counted for total population estimation were also sampled for cell volume estimation. Mean cell volume (MCV) was determined based on the following equation:
where l is the length of the line proportional to the area of the object (Mouton et al. 1994). Coefficients of error (CE) were calculated for MCV.
Statistical analysis
Statistical differences were determined using a Mann–Whitney non-parametric test of significance on the GraphPad InStat3 program (La Jolla, California, USA). The coefficient of variation (CV = SD/mean), presented in parentheses, was calculated for volume and neuronal number. The CE for the different measurements was calculated as \( \sqrt {{\text{meanCE}}^{2} } \).
Results
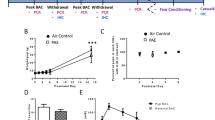
There was no significant difference found in terms of the average volume of the M layers between FAE (3.14 mm3, CV = 0.09) and control (2.76 mm3, CV = 0.14) subjects (P = 0.15). Likewise, the P regional volume did not differ between FAE (12.66 mm3, CV = 0.07) and control (12.14 mm3, CV = 0.17) subjects (P = 0.69; Fig. 1).

Cell populations a Volume estimation revealed no regional volume differences of the M and P regions between control and the FAE subjects. Number of neurons (b) and glia (c) in the M and P regions were not statistically different between control and FAE groups
The estimated total number of neurons in the M region (FAE = 374 × 103, CV = 0.107; Control = 338 × 103; CV = 0.097) was not statistically different between the two groups (P = 0.69; Fig. 2; Table 2). Neuronal estimations in the P region revealed no statistical difference between FAE (168 × 104, CV = 0.133) and control (163 × 104, CV = 0.087) subjects (P = 0.84; Fig. 1). The estimated total number of glia in the M region (FAE = 197 × 103, CV = 0.138; Control = 233 × 103; CV = 0.355) was not statistically different between the two groups (P = 0.69; Fig. 1). Glia estimations in the P region revealed no statistical difference between FAE (959 × 103, CV = 0.149) and control (991 × 103, CV = 0.216) subjects (P > 0.999; Fig. 1). The average CE for the number of glia was below 0.1 for both groups in the M and P-layers indicating an acceptable variation for this sampling scheme.

Soma volume the FAE group shows a 17% reduction in the M neuronal soma size compared to the control group and resembles the cell size typically seen in the P region. The size of the M-neurons of FAE subjects more closely resemble the size of neurons found in the P-layers. *P < 0.05
Average soma size was determined by randomly placing a standard grid size of 500 μm (x–y plane) for M and 750 μm (x–y plane) for P region on every 12th section through the LGN. At each intersection of the grid, an optical disector was positioned and an IUR nuclear rotator was placed on neurons that fell within the parameters of the disector were sampled. The average soma size of M neurons in FAE subjects (2,382.9 μm3; CV = 0.160), was significantly smaller than that of control subjects (2,856.8 μm3; CV = 0.068; P = 0.0287; Table 3; Fig. 2). The average soma size of P-neurons in FAE subjects (1,892.3 μm3; CV = 0.246), did not differ significantly from that of control subjects (2,222.6 μm3; CV = 0.18; P = 0.421; Fig. 3). Within the control group, the soma size of the P neurons is significantly smaller than that of M-neurons (P = 0.0159), whereas the soma size between these two neuronal types do not differ in the FAE group (P = 0.0823; Fig. 3).

Soma size differences are not visible at the global view of the whole LGN (a–d), M-neurons (shown by white arrows) of 3 representative FAE brains (f–h) appear to have smaller soma sizes than those of a representative normal brain (e) and are similar in size to the P-neurons (shown by black arrows; i–k). Scale bar = 1,250 µm (a–d); 50 µm (e–l)
Discussion
We have previously reported that the level of prenatal ethanol exposure described here results in a 35% neuronal reduction in the frontal cortex (Burke et al. 2009b) and a 59% neuronal reduction in the hippocampus (Burke et al. submitted). In the current study, FAE offspring demonstrate that there is no significant difference in the number of neurons, glia and in the overall laminar volume at birth in both regions of the LGN suggesting that the effect of prenatal ethanol varies depending on the brain region. However, the neuronal soma size in the M subdivision was significantly reduced by 17% compared to control values. The LGN neuronal and glia populations reported here for both groups resembles that previously reported in Macaca nemestrina and vervet monkeys (Blasco et al. 1999; Boire et al. 2002).
Rodent studies suggest that the thalamic areas are affected differently from other brain areas in response to prenatal ethanol exposure. Apoptotic proteins Bax, caspase-3 (apoptotic inducers), and Bcl-2 are (apoptotic inhibitor) are differentially expressed in the rodent brain following prenatal ethanol exposure. Throughout the cortex, prenatal ethanol exposure reduces Bcl-2 protein expression while elevating caspase-3 protein expression throughout the cortex (Mooney and Miller 2001). The level of Bax was unaffected but the ratio of Bcl-2/Bax cortical expression was significantly reduced following prenatal ethanol exposure. In contrast, Bax, Bcl-2, and caspase-3 protein expression are unaltered in thalamic nuclei with no change in Bcl-2/Bax ratio (Mooney and Miller 2001). Total neuronal population and volume of the thalamus are also unaltered in rodents exposed to prenatal ethanol during the second and third trimester equivalents (Livy et al. 2001) similar to that reported in the current study. Furthermore, layer IV of the visual cortex, the main recipient of thalamic inputs (Wise and Jones 1978; Frost and Caviness 1980), shows normal thickness and cell packing density following prenatal ethanol exposure (Miller and Dow-Edwards 1988; Miller and Potempa 1990). Rodent data suggest that the thalamus does not undergo a wave of programmed neuronal death induced by prenatal ethanol, as is the case with other brain areas (Mooney and Miller 2001).
The average soma size of M-neurons in ethanol-exposed subjects is smaller than that of control subjects. In addition, the soma sizes of M- and P-neurons in ethanol-exposed subjects are not significantly different from each other (Fig. 2). At around the 24th week of gestation in humans (approximately equal to the monkey embryonic day 100), there are no obvious differences between cells size in the M- and P- neurons (Hitchcock and Hickey 1980). This finding suggests that M-neurons of ethanol-exposed subjects have undergone slower-than-normal development and have been “frozen” in an embryonic state. One reason for the apparent “arrested development” of M-neurons in FAE subjects may be abnormal signalling during the gliogenesis period, which occurs during the last trimester of gestation. Prenatal ethanol exposure has been shown to impact the signalling of glia cells (Guerri et al. 2001) altering the levels of neurotrophins (Aronne et al. 2008; Moore et al. 2004). We have ruled out abnormal gliogenesis as a potential cause for the immature state of M-neurons. Since the M-neurons are the first to mature (Hitchcock and Hickey 1980), it stands to reason to conclude that the immature state of M-neurons of FAE subjects is a result of abnormal levels of neurotrophic factors.
The “arrested development” of the M-neurons may have far-reaching functional consequences with respect to the dorsal-stream visual processing. The output of the M- and P-LGN layers remains segregated in the primary visual cortex and leads to the emergence of two complementary streams of visual processing: the ventral stream, or the “what” pathway which leads to the representation of visual shape, form and identity of objects; and the dorsal stream, or the “where”/“how” pathway, which leads to the representation of visual space and visuomotor integration (for example see Mishkin et al. 1983; Milner and Goodale 1993).
The small, undifferentiated M-neurons of the FAS brain resemble a key neuroanatomical feature of fragile X syndrome (FXS), another neurodevelopmental disorder that is caused by the silencing of a single gene located on the X chromosome. Kogan and colleagues recently showed that the LGN of FXS brains lack the typical laminar pattern that differentiates M- from P-layers and that the entire LGN resembles an undifferentiated pool of neurons (Kogan et al. 2004). In addition, this neuroanatomical observation was found to be associated with a measureable visual functional impairment that affects only dorsal-stream visual processing. These impairments were measured in the form of an elevated threshold for motion coherence and lowering of both temporal and low-spatial contrast sensitivities, while ventral-stream related functions such as form coherence, chromatic and high-spatial contrast sensitivities remain spared. These careful observations lead us to believe that the small size of M-neurons following FAE will lead to a differential impact on visual processing such that dorsal-stream functions will be impaired while ventral-stream functions will remain normal.
There is also evidence from other forms of developmental intrusion or neurodevelopmental disorder that result in smaller-than-normal neuronal size accompanied by abnormal functional consequences. One line of evidence comes from sensory deprivation and/or anomalous sensory input throughout postnatal development. Monocular deprivation has been shown to bring about drastic reduction in the size of Y-cells in cat (Kageyama and Wong-Riley 1986; Ziburkus et al. 2000) and M-neurons in monkey (Tigges et al. 1984) LGN. Such anatomical modifications are coupled with abnormal functional consequences both at the single-neuron level (Levitt et al. 2001) and at the level of the entire LGN (Hess et al. 2009). In addition, evidence from the autism literature also suggests that the altered course of brain development in this emblematic form of neurodevelopmental disorder leads to smaller-than-normal neurons in the limbic system (Bauman and Kemper 1985; Raymond et al. 1996; Blatt et al. 2001; Bauman and Kemper 2005). This anatomical anomaly may in turn be the foundation for the social cognitive deficits observed in autism.
Breitmeyer and Ganz (1976) hypothesized that the M pathway could also play a direct role in directing selective attention during the reading task. This model proposes that the role of visual attention is to shift attention from the fixation point to the parafoveal region to the right of fixation so that the saccade may be programmed (Steinman et al. 1998). A shift in visual attention is necessary before a refixation may be executed (Sereno 1992), and the M pathway contributes an important input to the visual attention mechanisms (Steinman et al. 1997). These mechanisms also depend substantially upon neurons in the posterior parietal cortex, a major endpoint of the M pathway (Hussain 1991). The posterior parietal cortex has heavy connections with the frontal eye fields and the intermediate layers of the superior colliculus, both of which are important for saccadic eye movements (Mowafy et al. 1992). These studies propose that the M pathway may influence the planning of saccadic eye movements during reading tasks by means of an intermediate pathway (Steinman and Steinman 1998). A recent study has shown that children that were prenatally exposed to alcohol have impaired oculomotor control (Green et al. 2009), which maybe a consequence of an M pathway deficit.
Conclusion
The benefit of using the non-human as a translational model is that the naturalistic alcohol consumption can be monitored and thereby provides a controlled environment in which to study the effects of foetal alcohol exposure. The data presented here demonstrate that a regular and voluntarily consumption of alcohol during the third trimester, at doses that do not cause overt intoxication, does not affect the number of neurons in the P and M regions of the LGN. The soma size reduction of the M-neurons maybe related to an alcohol affect on neurotrophic factors released by astrocytes that would then have an effect on oculomotor control (Green et al. 2009). This study shows that not all regions of the brain are affected in the same manner by naturalistic third trimester ethanol exposure in primates (Burke et al. 2009b).
References
Archibald SL, Fennema-Notestine C, Gamst A, Riley EP, Mattson SN, Jernigan TL (2001) Brain dysmorphology in individuals with severe prenatal alcohol exposure. Dev Med Child Neurol 43:148–154
Aronne MP, Evrard SG, Mirochnic S, Brusco A (2008) Prenatal ethanol exposure reduces the expression of the transcriptional factor Pax6 in the developing rat brain. Ann NY Acad Sci 1139:478–498
Bauman M, Kemper TL (1985) Histoanatomic observations of the brain in early infantile autism. Neurology 35:866–874
Bauman ML, Kemper TL (2005) Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci 23:183–187
Blasco B, Avendano C, Cavada C (1999) A stereological analysis of the lateral geniculate nucleus in adult Macaca nemestrina monkeys. Vis Neurosci 16:933–941
Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML (2001) Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord 31:537–543
Boire D, Theoret H, Ptito M (2002) Stereological evaluation of neurons and glia in the monkey dorsal lateral geniculate nucleus following an early cerebral hemispherectomy. Exp Brain Res 142:208–220
Breitmeyer BG, Ganz L (1976) Implications of sustained and transient channels for theories of visual pattern masking, saccadic suppression, and information processing. Psychol Rev 83:1–36
Burke MW, Zangenehpour S, Mouton PR, Ptito M (2009a) Knowing what counts: unbiased stereology in the non-human primate brain. J Vis Exp (27):1262. doi:10.3791/1262
Burke MW, Palmour RM, Ervin FR, Ptito M (2009b) Neuronal reduction in frontal cortex of primates after prenatal alcohol exposure. Neuroreport 20:13–17
Burke MW, Zangenehpour S, Boire D, Ptito M (2009c) Dissecting the non-human primate brain in stereotaxic space. J Vis Exp (29):1–5. doi:10.3791/1259
Burke MW, Zangenehpour S, Ptito M (2009d) Brain banking: making the most of your research specimens. J Vis Exp (29):1260. doi:10.3791/1260
Clancy B, Darlington RB, Finlay BL (2001) Translating developmental time across mammalian species. Neuroscience 105:7–17
Clark CM, Li D, Conry J, Conry R, Loock C (2000) Structural and functional brain integrity of fetal alcohol syndrome in nonretarded cases. Pediatrics 105:1096–1099
Clarren SK, Astley SJ, Bowden DM, Lai H, Milam AH, Rudeen PK, Shoemaker WJ (1990) Neuroanatomic and neurochemical abnormalities in nonhuman primate infants exposed to weekly doses of ethanol during gestation. Alcohol Clin Exp Res 14:674–683
Colvin L, Payne J, Parsons D, Kurinczuk JJ, Bower C (2007) Alcohol consumption during pregnancy in nonindigenous west Australian women. Alcohol Clin Exp Res 31:276–284
Cook CS, Nowotny AZ, Sulik KK (1987) Fetal alcohol syndrome. Eye malformations in a mouse model. Arch Ophthalmol 105:1576–1581
Denny CH, Tsai J, Floyd RL, Green PP (2009) Alcohol use among pregnant and nonpregnant women of childbearing age—United States 1991–2005. Morbidity and mortality weekly report—centers for disease control, vol. 58, pp 529–532
Flanigan EY, Aros S, Bueno MF, Conley M, Troendle JF, Cassorla F, Mills JL (2008) Eye malformations in children with heavy alcohol exposure in utero. J Pediatr 153:391–395
Floyd RL, Sidhu JS (2004) Monitoring prenatal alcohol exposure. Am J Med Genet C Semin Med Genet 127C:3–9
Frost DO, Caviness VS Jr (1980) Radial organization of thalamic projections to the neocortex in the mouse. J Comp Neurol 194:369–393
Garey LJ, de Courten C (1983) Structural development of the lateral geniculate nucleus and visual cortex in monkey and man. Behav Brain Res 10:3–13
Green CR, Mihic AM, Brien DC, Armstrong IT, Nikkel SM, Stade BC, Rasmussen C, Munoz DP, Reynolds JN (2009) Oculomotor control in children with fetal alcohol spectrum disorders assessed using a mobile eye-tracking laboratory. Eur J Neurosci 29:1302–1309
Guerri C, Pascual M, Renau-Piqueras J (2001) Glia and fetal alcohol syndrome. Neurotoxicology 22:593–599
Gundersen HJ, Jensen EB (1987) The efficiency of systematic sampling in stereology and its prediction. J Microsc 147:229–263
Hess RF, Thompson B, Gole G, Mullen KT (2009) Deficient responses from the lateral geniculate nucleus in humans with amblyopia. Eur J Neurosci 29:1064–1070
Hitchcock PF, Hickey TL (1980) Prenatal development of the human lateral geniculate nucleus. J Comp Neurol 194:395–411
Hussain PF (1991) Visuospatial and visuomotor functions of the posterior parietal lobe. In: Stein JF (ed) Vision and visual dyslexia. CRC Press, Boca Raton, pp 12–43
Joelving FC, Billeskov R, Christensen JR, West M, Pakkenberg B (2006) Hippocampal neuron and glial cell numbers in Parkinson’s disease–a stereological study. Hippocampus 16:826–833
Kageyama GH, Wong-Riley M (1986) Differential effect of visual deprivation on cytochrome oxidase levels in major cell classes of the cat LGN. J Comp Neurol 246:212–237
Kogan CS, Boutet I, Cornish K, Zangenehpour S, Mullen KT, Holden JJ, Der Kaloustian VM, Andermann E, Chaudhuri A (2004) Differential impact of the FMR1 gene on visual processing in fragile X syndrome. Brain 127:591–601
Kristjanson AF, Wilsnack SC, Zvartau E, Tsoy M, Novikov B (2007) Alcohol use in pregnant and nonpregnant Russian women. Alcohol Clin Exp Res 31:299–307
Levitt JB, Schumer RA, Sherman SM, Spear PD, Movshon JA (2001) Visual response properties of neurons in the LGN of normally reared and visually deprived macaque monkeys. J Neurophysiol 85:2111–2129
Livy DJ, Maier Se, West JR (2001) Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the ventrolateral nucleus of the thalamus. Alcohol Clin Exp Res 25:774–780
Mattson SN, Schoenfeld AM, Riley EP (2001) Teratogenic effects of alcohol on brain and behavior. Alcohol Res Health 25:185–191
Miller MW, Dow-Edwards DL (1988) Structural and metabolic alterations in rat cerebral cortex induced by prenatal exposure to ethanol. Brain Res 474:316–326
Miller MW, Potempa G (1990) Numbers of neurons and glia in mature rat somatosensory cortex: effects of prenatal exposure to ethanol. J Comp Neurol 293:92–102
Milner AD, Goodale MA (1993) Visual pathways to perception and action. Prog Brain Res 95:317–337
Mishkin M, Ungerleider L, Macko K (1983) Object vision and spatial vision: two cortical pathways. Trends Neurosci 6:414–417
Mooney SM, Miller MW (2001) Effects of prenatal exposure to ethanol on the expression of bcl-2, bax and caspase 3 in the developing rat cerebral cortex and thalamus. Brain Res 911:71–81
Moore DB, Madorsky I, Paiva M, Barrow Heaton M (2004) Ethanol exposure alters neurotrophin receptor expression in the rat central nervous system: effects of neonatal exposure. J Neurobiol 60:114–126
Mouton PR, Pakkenberg B, Gundersen HJ, Price DL (1994) Absolute number and size of pigmented locus coeruleus neurons in young and aged individuals. J Chem Neuroanat 7:185–190
Mowafy L, Lappin JS, Anderson BL, Mauk DL (1992) Temporal factors in the discrimination of coherent motion. Percept Psychophys 52:508–518
Paxinos G, Huang XF, Toga AW (1999) Rhesus monkey brain in stereotaxic coordinates. Elsevier & Academic Press, Amsterdam
Raymond GV, Bauman ML, Kemper TL (1996) Hippocampus in autism: a Golgi analysis. Acta Neuropathol 91:117–119
Riley EP, McGee CL (2005) Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood) 230:357–365
Riley EP, McGee CL, Sowell ER (2004) Teratogenic effects of alcohol: a decade of brain imaging. Am J Med Genet C Semin Med Genet 127C:35–41
Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, Hanson JW, Graham JM Jr (1997) Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology 56:317–326
Sereno AB (1992) Programming saccades: the role of attention. In: Rayner K (ed) Eye movements and visual cognition: Scene perception and reading. Springer, New York, pp 89–107
Sidhu JS, Floyd RL (2002) Alcohol use among women of childbearing age—United States, 1991–1999. Morbidity and mortality weekly report—centers for disease control, vol. 51, pp 273–276
Spadoni AD, McGee CL, Fryer SL, Riley EP (2007) Neuroimaging and fetal alcohol spectrum disorders. Neurosci Biobehav Rev 31:239–245
Steinman SB, Steinman BA (1998) Vision and attention. I: Current models of visual attention. Optom Vis Sci 75:146–155
Steinman BA, Steinman SB, Lehmkuhle S (1997) Transient visual attention is dominated by the magnocellular stream. Vision Res 37:17–23
Steinman SB, Steinman BA, Garzia RP (1998) Vision and attention. II: Is visual attention a mechanism through which a deficient magnocellular pathway might cause reading disability? Optom Vis Sci 75:674–681
Streissguth AP, Sampson PD, Olson HC, Bookstein FL, Barr HM, Scott M, Feldman J, Mirsky AF (1994) Maternal drinking during pregnancy: attention and short-term memory in 14-year-old offspring–a longitudinal prospective study. Alcohol Clin Exp Res 18:202–218
Stromland K (1985) Ocular abnormalities in the fetal alcohol syndrome. Acta Ophthalmol Suppl 171:1–50
Stromland K, Pinazo-Duran MD (2002) Ophthalmic involvement in the fetal alcohol syndrome: clinical and animal model studies. Alcohol Alcohol 37:2–8
Tenkova T, Young C, Dikranian K, Labruyere J, Olney JW (2003) Ethanol-induced apoptosis in the developing visual system during synaptogenesis. Invest Ophthalmol Vis Sci 44:2809–2817
Tigges M, Hendrickson AE, Tigges J (1984) Anatomical consequences of long-term monocular eyelid closure on lateral geniculate nucleus and striate cortex in squirrel monkey. J Comp Neurol 227:1–13
Tsai J, Floyd RL (2004) Alcohol consumption among women who are pregnant or who might become pregnant—United States, 2002. Morbidity and mortality weekly report—centers for disease control, vol. 53, pp 1178–1181
West MJ, Gundersen HJ (1990) Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 296:1–22
West MJ, Slomianka L, Gundersen HJ (1991) Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231:482–497
Wise SP, Jones EG (1978) Developmental studies of thalamocortical and commissural connections in the rat somatic sensory cortex. J Comp Neurol 178:187–208
Ziburkus J, Bickford ME, Guido W (2000) NMDAR-1 staining in the lateral geniculate nucleus of normal and visually deprived cats. Vis Neurosci 17:187–196
Author information
Authors and Affiliations
Corresponding author
Additional information
M. F. Papia and M. W. Burke are co-first authors.
Rights and permissions
About this article
Cite this article
Papia, M.F., Burke, M.W., Zangenehpour, S. et al. Reduced soma size of the M-neurons in the lateral geniculate nucleus following foetal alcohol exposure in non-human primates. Exp Brain Res 205, 263–271 (2010). https://doi.org/10.1007/s00221-010-2364-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-010-2364-6