
Abstract
Chromatin modification plays a key role in fate decision of neural stem cells. Here, we explored the impact of epigenetic remodelling onto neuronal fate determination using specific inhibitors of histone deacetylases (iHDAC). Adult subventricular zone (SVZ) precursor cells were expanded as neurospheres and treated in vitro with second generation iHDAC MS-275, M344 and suberoylanilide hydroxamic acid (SAHA). All tested compounds revealed a significant increase of βIII-tubulin positive neurons (ranging from 258 to 431%) in a concentration-dependent manner. The number of oligodendrocytes was decreased by almost 50%, accompanied by a reduction of Olig2 mRNA expression. In contrast, astrocyte quantity remained unaffected after iHDAC treatment. Both control and iHDAC treated cells expressed markers of mature GABAergic and dopaminergic neurons. Increased expression levels of NeuroD, Cyclin D2 and B-lymphocyte translocation gene 3 (Btg3) point to a shift towards neuronal fate determination targeted by HDAC inhibitors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adult stem cells of the central nervous system (CNS) persist in the SVZ as well as in the hippocampal dentate gyrus (Ming and Song 2005). These cells have the ability to differentiate into glial or neuronal cell progenies, i.e., astrocytes, oligodendrocytes or neurons. Recent data point to epigenetic regulatory mechanisms as important internal signals for cell lineage determination. Epigenetic regulation of gene expression involves posttranslational modification of N-terminal histone residues, i.e., acetylation, deacetylation and methylation of conserved lysines. Acetylation at lysine residues is associated with transcriptional activation, while deacetylation followed by methylation confers gene silencing (Peterson and Laniel 2004). Deacetylation is rendered by histone deacetylases (HDACs) which are components of large chromatin silencing complexes including cofactors such as neuron restrictive silencer factor (NRSF), also known as RE1 silencing transcription factor (REST) (Naruse et al. 1999). Pharmacological inhibition of HDAC enzymes enables experimental manipulation and systematic analysis of chromatin remodelling (Marks et al. 2003). HDAC inhibition leads to excessive histone acetylation and transcriptional activation. Amongst the large number of differentially expressed genes, HDAC inhibitors were found to upregulate neuronal transcription factors, such as NeuroD (Hsieh et al. 2004). Additionally, the cell cycle regulator Cyclin D2 emerged as important candidate gene (Laeng et al. 2004). Previous studies showed the antiepileptic drug valproic acid (VPA) to modulate neuronal differentiation of stem cells (Hsieh et al. 2004; Laeng et al. 2004). As there are multiple modes of action known for VPA and rather large concentrations in the millimolar range are needed to inhibit HDACs, some controversy remains with respect to the exact mechanism by which neuronal differentiation was induced (Hao et al. 2004). Here, we examined the effects of highly specific second generation HDAC inhibitors, such as SAHA, MS-275 and M344 on neuroepithelial lineage determination of adult SVZ-derived neural precursor cells (NPCs). Hydroxamic acids SAHA and M344 are highly selective for HDAC class I and II isoenzymes and operate already at low micromolar concentrations (Miller et al. 2003). In contrast, the benzamide MS-275 interacts at low concentrations only with HDAC class I enzymes (Park et al. 2004; Hahnen et al. 2006).
Results
Neural precursor cells (NPC) were isolated from adult SVZ and formed neurospheres in culture, which could be continuously passaged and proved multipotent. Sphere-derived cells were able to clonally form neurospheres when sorted as single cells into 96-well plates (limiting dilution assay). Clonal spheres were also able to form secondary spheres upon dissociation and replating (data not shown). Only neurospheres after passage two were used in subsequent experiments.
To test the effect of HDAC inhibitors on NPC differentiation, neurosphere-derived cells were differentiated by growth factor withdrawal and serum addition. One hour after plating, HDAC inhibitors SAHA, MS-275 and M344 or DMSO vehicle were added to the cultures. The potency of SAHA to affect histone acetylation was confirmed by immunoblotting for Lys-9 acetylated histone H3 (not shown).
Numbers of differentiated neurons were assessed by double-immunostaining for βIII tubulin and GFAP. All tested compounds were able to significantly enhance neuronal differentiation of cultured NPCs. Total as well as immunocytochemically identified neuronal cell numbers were counted in ten randomly placed visual fields (0.6 mm2). Total cell numbers were within the same range for compound and vehicle treated experiments (data not shown).
To examine whether fate modulation was concentration-dependent, neurospheres were differentiated for seven days with MS-275, SAHA or M344 in concentrations ranging from 0.1 to 2 μM. All compounds increased neuronal differentiation with peak numbers at following concentrations: 0.5 μM SAHA increased neuronal differentiation by 285%, 1 μM MS-275 increased neuronal differentiation by 273% and 1 μM M344 increased neuronal differentiation by 226%, respectively. In contrast, reduced cell numbers were observed at concentrations of >1 μM of SAHA, M344 and MS-275.
After analysis of different iHDAC concentrations, we tested whether short-term exposure to iHDAC was sufficient to induce neuronal fate determination. HDAC inhibitors SAHA (0.5 μM), MS-275 (1 μM) and M344 (1 μM) were added to differentiating neurosphere-derived cells. Inhibitors were washed out and medium was exchanged after 24 and 48 h, respectively. The most pronounced increase in neuronal differentiation was found after 24 h of treatment with M344 (295%) and MS-275 (258%) and after 48 h with SAHA (431%; Fig. 1a). This increase was found to be more prominent than long-term exposure for 7 days.

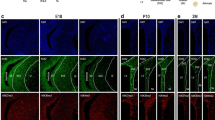
Effect of HDAC inhibitor treatment on neurosphere differentiation. a Treatment of neurosphere-derived cells with MS-275, M344 or SAHA leads to a concentration-dependent induction of neuronal differentiation. NPCs were treated with 0.5 μM SAHA (48 h), 1 μM MS-275 (24 h), 1 μM M344 (24 h) or vehicle. Differentiated cells were analysed by βIII tubulin, GFAP and CNPase immunostaining. Given are distributions of cell types after treatment with indicated compounds ± SEM. Treatment induced no significant changes in astrocyte numbers, while oligodendrocytes decreased. All experiments were carried out in triplicate with at least 2,000 cells per experiment. Asterisks indicate statistical significance (Student’s T-Test, P < 0.001). b Propidium iodide incorporation assay. NPCs were treated with HDAC inhibitors and exposed to 5 μg/ml propidium iodide after 24 and 48 h, respectively. Given are percentages of propidium iodide positive cells of the total cell population. All experiments were carried out in triplicate with asterisks indicating statistical significance (Student’s T-Test; *P < 0.05; **P < 0.01). c Map2-immunostaining of DMSO vehicle and SAHA (0.5 μM, 48 h) treated neurosphere derived cells. Quantification of this cell population showed a significant increase of Map2 expressing cells in SAHA-treated experiments (n > 2000 cells counted). Asterisks indicate statistical significance (P < 0.001) compared to controls. Scale bar 20 μm. d Neurosphere derived DMSO vehicle or SAHA (0.5 μM, 48 h) treated cells were differentiated for 28 days and subsequently stained for markers of GABAergic or dopaminergic neurons. Controls and iHDAC treated cells expressed markers for both populations (GAD65/67 and TH) in combination with βIII tubulin. Scale bar 10 μm
To analyse toxicity of the used compounds, we performed a propidium iodide incorporation assay. Cell death was examined by the incorporation of propidium iodide and assessed after 24 and 48 h, respectively. Numbers of propidium iodide positive cells were quantified and the ratio of propidium iodide positive cells versus total cells calculated. None of the used substances induced cell death at low concentrations (0.5–1 μM). However, higher concentrations (2–5 μM) significantly increased propidium iodide incorporation (Fig. 1b), compatible with enhanced cell death (Marks and Jiang 2005).
To analyse whether fate determination of other cell lineages was influenced by HDAC inhibitors, we performed double-immunofluorescence analysis. Neurosphere-derived cells were differentiated for seven days and treated with HDAC inhibitors or vehicle and subsequently immunostained either with a combination of anti-ßIII tubulin and anti-GFAP or anti-GFAP and anti-CNPase antibodies. All tested compounds increased the number of newly formed neurons (by 258%–431%), while numbers of oligodendrocytes decreased (P < 0.001, T-test). The astrocytic cell population characterized by GFAP immunoreactivity remained unaffected (Fig. 1a).
We further analysed the neuronal phenotype of vehicle (DMSO, 48 h) or SAHA (0.5 μM, 48 h) treated cells using Map2 immunocytochemisty. SAHA treatment increased the number of Map2 expressing cells compared to controls (DMSO) by 185% (Fig. 1c). Cells were allowed to differentiate for 7 days. In addition, prolonged differentiation periods of 28 days allowed identifying neurons with a GABAergic (GAD65/67) or dopaminergic (TH) phenotype. These markers colocalized with βIII tubulin expression (Fig. 1d). Although we have not performed a quantitative analysis, differentiation capability in long-term cultures was not compromised by iHDAC treatment.
As shown by quantitative real-time PCR, the activating transcription factor 5 (Atf5) was downregulated significantly following iHDAC treatment. Atf5 has been implicated in maintenance of stem cell proliferation and Atf5 expression levels decrease with onset of differentiation (Angelastro et al. 2003; Mason et al. 2005). Previous reports pointed to the transcription factor NeuroD and the cell-cycle control gene Cyclin D2 to be upregulated by HDAC inhibitors (Hsieh et al. 2004; Laeng et al. 2004). We confirmed these findings by quantitative RT-PCR analysis in our neurosphere culture approach. Furthermore, expression of Cyclin D3 was unaffected (not shown) while Cyclin D1 was downregulated. We identified a novel gene induced by HDAC inhibition. SAHA, M344 and MS-275 were able to up-regulate the B-lymphocyte translocation gene 3 (Btg3) in neurosphere cultures (Fig. 2). Btg3 is a cell-cycle inhibitory gene and belongs to the PC3/Btg/TOB family of antiproliferative genes (Tirone 2001). Another member of this family (Btg2) previously identified in neuronal development (Canzoniere et al. 2004) was not affected by HDAC inhibitor treatment. In contrast to increased levels of neuronal differentiation markers, we identified significantly lower mRNA expression of oligodendrocyte lineage transcription factor 2 (Olig2). The downregulation of Olig2 is in accordance with decreased numbers of CNPase-immunoreactive oligodendrocytes (Figs. 1a, 2).

Real-time PCR of candidate genes for HDAC inhibitor induced differentiation. NPCs were treated with 0.5 μM SAHA (48 h), 1 μM MS-275 (24 h), 1 μM M344 (24 h) or vehicle. Quantitative real time RT-PCR for NeuroD1, Cyclin D2 and Btg3 revealed significant induction by HDAC inhibitors, while expression of Atf5, Cyclin D1 and Olig2 was decreased. Given are relative expression levels compared to GAPDH. Asterisks indicate levels of statistical significance (one-way ANOVA, *P < 0.05; **P < 0.01; ***P < 0.001)
Discussion
A pool of dormant stem cells survives in restricted niches of the CNS throughout life (Gage 2000) and may be a valuable resource for tissue regeneration. However, it is mandatory to elucidate molecular mechanisms of recruitment, proliferation and differentiation of adult brain stem cells in order to generate sufficient amounts of distinct cellular subpopulations. Here, we tested the propensity of HDAC inhibitors to pharmacologically induce neuronal fate determination from postnatal precursor cells. Self-renewal properties in our paradigm were systematically examined testing the capacity of clonally derived neurospheres to form secondary spheres in vitro. Furthermore, self-renewing neurospheres had the potency to generate glial and neuronal progenies after growth factor withdrawal and serum addition. Following incubation with specific HDAC inhibitors neuronal differentiation was induced by 258–431% (Fig. 1a). The non-specific compound VPA was more effective in inducing neuronal differentiation in other studies. Whether this is due to other VPA mediated effect, e.g., PI3 kinase inhibition remains to be specified.
While SAHA and M344 are non-selective inhibitors of histone deacetylases, restricted specificity of MS-275 for the class I enzymes HDAC1 and 2 (Park et al. 2004) points to targeted recruitment of transcriptional complexes during cell lineage determination. Interestingly, restriction of pro-neuronal genes by the NRSF/mSin3/HDAC1 complex has been reported (Naruse et al. 1999). We propose a molecular pathway for neuronal fate determination in which inhibition of the NRSF/mSin3/HDAC1 complex promotes neuronal differentiation from neural NPCs. On the other hand, Trichostatin A appeared to prevent oligodendrogenesis indicating that lineage determination of oligodendrocytes is also dependent on HDAC activity (Marin-Husstege et al. 2002). This observation is confirmed by our analysis using second-generation iHDAC.
Astrocyte populations were not affected in our experimental approach, although previous studies showed a significant reduction of the astrocytic population after treatment with VPA (Hsieh et al. 2004; Laeng et al. 2004). Several possibilities should be considered. Inconsistencies obtained from different culture models (regarding both origin of used cells, age and/or strain of used animals) may result from variable numbers of endogenous astrocytic progenitors. In addition, we have not quantified those cells coexpressing βIII tubulin and GFAP, which may contribute to different behaviour of astrocytic cell populations reported in other studies (Hsieh et al. 2004).
We tested the ability of iHDAC treated cells for long-term differentiation (28 days) (Fig. 1d). Both controls and SAHA treated cells showed features of GABAergic (GAD65/76) and dopaminergic (TH) differentiation, while none of the cells expressed markers for glutamatergic neurons (vGlut1, data not shown), thus indicating that iHDAC do not induce a posterior fate from anterior precursor cells. Interestingly, a regulation of the TH promoter by NRSF has been described recently (Kim et al. 2006). NRSF is able to form a complex with HDAC1, which may be disrupted by iHDAC, thus promoting dopaminergic differentiation.
We were further interested to identify candidate genes associated with differentiation effects of iHDAC and analysed, therefore, mRNA expression levels of Atf 5, Cyclin D1, D2 and D3, NeuroD1, Btg3 and Olig2 genes (Fig. 2). We observed a downregulation of Atf5, which has been reported as prerequisite for induction of differentiation in neural stem/progenitor cells (Angelastro et al. 2003; Mason et al. 2005). Upregulation of NeuroD1 and Cyclin D2 was implicated in inducing neuronal differentiation using VPA (Hsieh et al. 2004; Laeng et al. 2004). Importantly, we confirmed these findings in our assay. Of the other D-type cyclins, Cyclin D3 remained unchanged while Cyclin D1 was significantly downregulated. Different functions for the three D-type cyclins have been shown and cytoplasmic sequestration of Cyclin D1 is known to enhance neuronal survival (Sumrejkanchanakij et al. 2003; Baker et al. 2005). Genetic ablation of Cyclin D1 does not influence neurogenesis during development while a Cyclin D2 knockout results in complete loss of proliferating progenitor cells (Kowalczyk et al. 2004). It is conceivable from these studies that (i) Cyclin D2 promotes proliferation of early neuronal progenitors and that (ii) downregulation or nuclear depletion of Cyclin D1 protects newborn neurons from apoptosis.
Moreover, we identified Btg3 as promising new target gene for neuronal differentiation in neural precursor cells. Btg3 was prominently upregulated in neural precursor cells after iHDAC treatment. Btg3 is a cell-cycle inhibitor and belongs to the PC3/Btg/TOB family of antiproliferative genes (Tirone 2001). Interestingly, Btg3 is expressed in neurogenic regions of the developing CNS (Yoshida et al. 1998) and other members of this protein family (i.e., Btg2) are capable to induce neuronal differentiation (Canzoniere et al. 2004). However, Btg2 is not regulated in our experimental assay but has the ability to downregulate Cyclin D1 expression (Guardavaccaro et al. 2000). Whether this also applies to Btg3 remains to be shown.
We also found a distinct downregulation of Olig2. Olig2 is a basic helix-loop-helix transcription factor expressed in those subventricular zone stem cells differentiating into oligodendrocyte lineage (Ligon et al. 2006). Overexpression of Olig2 in adult SVZ cells leads to a distinct increase of oligodendroglial cell numbers in vivo (Hack et al. 2005). In our experiments, reduction of oligodendrocyte differentiation most likely results from a downregulation of Olig2 by iHDAC (Fig. 2). Furthermore, dependency of oligodendrogenesis on the HDAC1 enzyme activity has been recently confirmed in zebrafish (Cunliffe and Casaccia-Bonnefil 2006).
In conclusion, inhibition of HDAC complexes in SVZ precursor cells induced a shift in fate decision from oligodendrocytes to neurons. In vivo, “type C cells” of the SVZ (Doetsch et al. 1997) either differentiate into neuronal cells (upon expression of proneural genes, such as NeuroD) or into oligodendrocytes (when expressing oligodendrocyte inducing genes, such as Olig2). Decision of which genes are expressed largely depends on transcriptional complexes and recruitment of HDAC enzymes, with HDAC1 being the most important protein (Naruse et al. 1999; Cunliffe and Casaccia-Bonnefil 2006). Hence, selective iHDAC are useful tools to experimentally induce neuronal fate decision in SVZ precursor cells.
Material and methods
Suberoylanilide hydroxamic acid (SAHA) was obtained from Alexis (Grünberg, Germany), N-(2-aminophenyl)-4-[N-(pyridin-3-yl-methoxycarbonyl)aminomethyl] benzamide (MS-275) and N-Hydroxy-7-(4-dimethylaminobenzoyl)aminoheptanamide (M344) from Calbiochem (Darmstadt, Germany).
Isolation of SVZ cells from postnatal day six Wistar rats was performed as described previously (Wachs et al. 2003). All animals were handled according to the “Principles of laboratory animal care” (NIH publication No. 86–23, revised 1985) and the German Law on the Protection of Animals. Primary cells were plated into six-well culture plates with N2 medium containing DMEM/F-12 (1:1, Invitrogen, Karlsruhe, Germany), 0.1 g/l penicillin/streptomycin (Sigma, Taufkirchen, Germany), 2 mM l-glutamine, 5 μg insulin (Chemicon, Chandlers Ford, UK), 100 μg transferrin (Chemicon), 100 μM putrescine (Sigma), 30 nM sodium selenite (Sigma), 20 nM progesterone (Sigma) and 20 ng/ml EGF and bFGF (r&d systems, Minneapolis, USA). Neurospheres were passaged using Accutase (PAA, Cölbe, Germany) according to the manufacturer’s protocol. For differentiation analysis 50,000 cells were plated onto laminin (Tebu Bio, Offenbach, Germany) and poly-l-ornithine (Sigma) coated coverslips in N2 medium with 10% fetal bovine serum (Biochrom, Berlin, Germany) and treated with HDAC inhibitors where indicated. Differentiated cells were fixed with 4% paraformaldehyde.
For limiting dilution analysis, neurospheres were dissociated and separated by a Mo-Flo cell sorter (Dako Cytomation, Glostrup, Denmark). Medium exchange was performed every second week.
Immunostaining of fixed cells was performed as described elsewhere (Wachs et al. 2003). Primary antibodies were used in the following dilutions: Mouse anti-βIII tubulin (Promega, Mannheim, Germany, 1:2,000), rabbit anti-glial fibrillary acidic protein (GFAP, Dako, Denmark, 1:1000) mouse anti-2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase, Chemicon, USA, 1:200), mouse anti-microtubule associated protein 2 (Map2, clone C, a kind gift of Dr. B. Riederer, Lausanne, 1:25), rabbit anti-tyrosine hydroxylase (TH, Chemicon, 1:200) and rabbit anti-glutamic acid decarboxylase (GAD65/67, Chemicon, 1:200). Secondary antibodies were used in 1:250 dilutions (Goat anti-mouse Cy3 and Goat anti-rabbit Cy2, Dianova, Germany). Hoechst 33258 (500 ng/ml, Sigma) was used for nuclear counterstaining.
Propidium iodide incorporation assay was performed as described previously (Eyupoglu et al. 2005). Briefly, propidium iodide (Sigma) was added to the culture medium of differentiating cells at a final concentration of 5 μg/ml. Following incubation for 15 min at 37°C in the dark, cells were washed once with PBS and incubated in fresh medium for 5 min at 37°C in the dark. The medium was exchanged again prior to fluorescence microscopical analysis.
Qualitative and quantitative analysis of immunocytochemically identified cell types was performed using high power optical fields digitized with a CCD camera (F View II, Soft Imaging Systems, Münster, Germany) equipped to an Olympus BX51 microscope (Olympus, Hamburg, Germany) and respective imaging software (analySIS, Soft Imaging Systems). Nuclear counterstaining was quantified in all experiments and used as reference value to assess the percentage of neuronal and glial subpopulations. Statistical significance was calculated using Student’s T-Test (GraphPad Prism 4.02, GraphPad Software, USA).
For quantitative gene expression analysis, NPCs from the SVZ of postnatal day six C57/Bl6 mice were cultured and differentiated according to the above-mentioned protocol. For relative quantification of target gene expression, one step RT-PCR was preformed using the QuantiTect SYBR Green RT-PCR Kit and validated QuantiTect Primer Assays (QIAGEN, Hilden, Germany) on an Applied Biosystems 7500 Real-Time PCR System. The comparative method of relative quantification (2−ΔΔCt) was used to calculate expression levels of each target gene (normalized to GAPDH) and compared to the non-treated control samples.
Protein extraction and western blot of acetylated histones was performed as described previously (Eyupoglu et al. 2005).
References
Angelastro JM, Ignatova TN, Kukekov VG, Steindler DA, Stengren GB, Mendelsohn C, Greene LA (2003) Regulated expression of ATF5 is required for the progression of neural progenitor cells to neurons. J Neurosci 23:4590–4600
Baker GL, Landis MW, Hinds PW (2005) Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle 4:330–338
Canzoniere D, Farioli-Vecchioli S, Conti F, Ciotti MT, Tata AM, Augusti-Tocco G, Mattei E, Lakshmana MK, Krizhanovsky V, Reeves SA, Giovannoni R, Castano F, Servadio A, Ben-Arie N, Tirone F (2004) Dual control of neurogenesis by PC3 through cell cycle inhibition and induction of Math1. J Neurosci 24:3355–3369
Cunliffe VT, Casaccia-Bonnefil P (2006) Histone deacetylase 1 is essential for oligodendrocyte specification in the zebrafish CNS. Mech Dev 123:24–30
Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A (1997) Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 17:5046–5061
Eyupoglu IY, Hahnen E, Buslei R, Siebzehnrubl FA, Savaskan NE, Luders M, Trankle C, Wick W, Weller M, Fahlbusch R, Blumcke I (2005) Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J Neurochem 93:992–999
Gage FH (2000) Mammalian neural stem cells. Science 287:1433–1438
Guardavaccaro D, Corrente G, Covone F, Micheli L, D’Agnano I, Starace G, Caruso M, Tirone F (2000) Arrest of G(1)-S progression by the p53-inducible gene PC3 is Rb dependent and relies on the inhibition of cyclin D1 transcription. Mol Cell Biol 20:1797–1815
Hack MA, Saghatelyan A, de Chevigny A, Pfeifer A, Ashery-Padan R, Lledo PM, Gotz M (2005) Neuronal fate determinants of adult olfactory bulb neurogenesis. Nat Neurosci 8:865–872
Hahnen E, Eyupoglu IY, Brichta L, Haastert K, Trankle C, Siebzehnrubl FA, Riessland M, Holker I, Claus P, Romstock J, Buslei R, Wirth B, Blumcke I (2006) In vitro and ex vivo evaluation of second generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J Neurochem 98:193–202
Hao Y, Creson T, Zhang L, Li P, Du F, Yuan P, Gould TD, Manji HK, Chen G (2004) Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J Neurosci 24:6590–6599
Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH (2004) Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci USA 101:16659–16664
Kim SM, Yang JW, Park MJ, Lee JK, Kim SU, Lee YS, Lee MA (2006) Regulation of human tyrosine hydroxylase gene by neuron-restrictive silencer factor. Biochem Biophys Res Commun 346:426–435
Kowalczyk A, Filipkowski RK, Rylski M, Wilczynski GM, Konopacki FA, Jaworski J, Ciemerych MA, Sicinski P, Kaczmarek L (2004) The critical role of cyclin D2 in adult neurogenesis. J Cell Biol 167:209–213
Laeng P, Pitts RL, Lemire AL, Drabik CE, Weiner A, Tang H, Thyagarajan R, Mallon BS, Altar CA (2004) The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J Neurochem 91:238–251
Ligon KL, Fancy SP, Franklin RJ, Rowitch DH (2006) Olig gene function in CNS development and disease. Glia 54:1–10
Marin-Husstege M, Muggironi M, Liu A, Casaccia-Bonnefil P (2002) Histone deacetylase activity is necessary for oligodendrocyte lineage progression. J Neurosci 22:10333–10345
Marks PA, Jiang X (2005) Histone deacetylase inhibitors in programmed cell death and cancer therapy. Cell Cycle 4:549–551
Marks PA, Miller T, Richon VM (2003) Histone deacetylases. Curr Opin Pharmacol 3:344–351
Mason JL, Angelastro JM, Ignatova TN, Kukekov VG, Lin G, Greene LA, Goldman JE (2005) ATF5 regulates the proliferation and differentiation of oligodendrocytes. Mol Cell Neurosci 29:372–380
Miller TA, Witter DJ, Belvedere S (2003) Histone deacetylase inhibitors. J Med Chem 46:5097–5116
Ming GL, Song H (2005) Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci 28:223–250
Naruse Y, Aoki T, Kojima T, Mori N (1999) Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron-specific target genes. Proc Natl Acad Sci USA 96:13691–13696
Park JH, Jung Y, Kim TY, Kim SG, Jong HS, Lee JW, Kim DK, Lee JS, Kim NK, Bang YJ (2004) Class I histone deacetylase-selective novel synthetic inhibitors potently inhibit human tumor proliferation. Clin Cancer Res 10:5271–5281
Peterson CL, Laniel MA (2004) Histones and histone modifications. Curr Biol 14:R546–551
Sumrejkanchanakij P, Tamamori-Adachi M, Matsunaga Y, Eto K, Ikeda MA (2003) Role of cyclin D1 cytoplasmic sequestration in the survival of postmitotic neurons. Oncogene 22:8723–8730
Tirone F (2001) The gene PC3(TIS21/BTG2), prototype member of the PC3/BTG/TOB family: regulator in control of cell growth, differentiation, and DNA repair? J Cell Physiol 187:155–165
Wachs FP, Couillard-Despres S, Engelhardt M, Wilhelm D, Ploetz S, Vroemen M, Kaesbauer J, Uyanik G, Klucken J, Karl C, Tebbing J, Svendsen C, Weidner N, Kuhn HG, Winkler J, Aigner L (2003) High efficacy of clonal growth and expansion of adult neural stem cells. Lab Invest 83:949–962
Yoshida Y, Matsuda S, Ikematsu N, Kawamura-Tsuzuku J, Inazawa J, Umemori H, Yamamoto T (1998) ANA, a novel member of Tob/BTG1 family, is expressed in the ventricular zone of the developing central nervous system. Oncogene 16:2687–2693
Acknowledgments
The authors thank D. Müller for excellent technical assistance. This work has been supported by the Bavarian Research Council (Munich) and the Johannes and Frieda Marohn Foundation (University of Erlangen). FAS is a fellow of the Studienstiftung des deutschen Volkes e.V. (German national merit foundation).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Siebzehnrubl, F.A., Buslei, R., Eyupoglu, I.Y. et al. Histone deacetylase inhibitors increase neuronal differentiation in adult forebrain precursor cells. Exp Brain Res 176, 672–678 (2007). https://doi.org/10.1007/s00221-006-0831-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-006-0831-x