
Abstract
The role of the neutral amino acid glycine in excitotoxic neuronal injury is unclear. Glycine coactivates glutamate N-methyl-D-aspartate (NMDA) receptors by binding to a distinct recognition site on the NR1 subunit. Purely excitatory glycine receptors composed of NR1 and NR3/NR4 NMDA receptor subunits have recently been described, raising the possibility of excitotoxic effects mediated by glycine alone. We have previously shown that exposure to high concentrations of glycine causes extensive neurotoxicity in organotypic hippocampal slice cultures by activation of NMDA receptors. In the present study, we investigated further properties of in vitro glycine-mediated toxicity. Agonists of the glycine recognition site of NMDA receptors (D-serine and D-alanine) did not have any toxic effect in hippocampal cultures, whereas competitive blockade of the glycine site by 7-chlorokynurenic acid was neuroprotective. Stimulation (taurine, β-alanine) or inhibition (strychnine) of the inhibitory strychnine-sensitive glycine receptors did not produce any neurotoxicity. The toxic effects of high-dose glycine were comparable in extent to those produced by the excitatory amino acid glutamate in our model. When combined with sublethal hypoxia/hypoglycemia, the threshold of glycine toxicity was decreased to less than 1 mM, which corresponds to the range of concentrations of excitatory amino acids measured during in vivo cerebral ischemia. Taken together, these results further support the assumption of an active role of glycine in excitotoxic neuronal injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Excitatory amino acid-mediated toxicity has been recognized as a major cause of neuronal cell death in different neurologic diseases such as ischemia, epilepsy, and trauma (Choi and Rothman 1990; Lipton and Rosenberg 1994). Extracellular release of glutamate after acute brain injury leads to uncontrolled activation of N-methyl-D-aspartate (NMDA) receptors, intracellular penetration of calcium, and initiation of deleterious processes finally leading to cell death. The role of the neutral amino acid glycine in excitotoxic neuronal injury is less clear. Under physiologic conditions, glycine is a necessary coactivator of NMDA receptors by binding a site distinct from the glutamate recognition site and by allosterically promoting glutamate activation of the NMDA-associated ion channel (Johnson and Ascher 1987; Kleckner and Dingledine 1988; Thomson 1990; Dingledine et al. 1999; Foucaud et al. 2003). Several older studies have reported that glycine alone may be able to activate NMDA receptors in the absence of glutamate or NMDA (Meguro et al. 1992; Kutsawada et al. 1992; Pace et al. 1992). In a recent study using subunit recombinant methods, glycine was found to fully and exclusively activate NMDA receptors containing the NR3 subunit family (Chatterton et al. 2002). This suggests the presence of functional excitatory glycine receptors in the brain.
Abnormal elevations of glycine concentration accompanying that of excitatory amino acids have been reported in several pathologic conditions, supporting the assumption that glycine may also play a role in excitotoxic neuronal injury (Perry and Hansen 1981; Larson and Beitz 1988; Globus et al. 1991a, 1991b; Kanthan and Shuaib 1995; Castillo et al. 1996; Saransaari and Oja 2001). In acutely prepared hippocampal slices, exogenously applied high concentrations of glycine caused electrophysiological manifestations of excitotoxicity similar to the effects of glutamate (Wallis et al. 1995). Whole cell voltage-clamp recordings of cultured rat hippocampal neurons showed that high concentrations of glycine and other neutral amino acids were able to elicit inward current responses by activation of NMDA receptors (Pace et al. 1992). In a previous study, we have found that high concentrations of glycine (2–4 mM) caused hyperexcitability and subsequent neuronal cell death in rat organotypic hippocampal slice cultures (Newell et al. 1997). These neurotoxic effects occurred in all regions of the hippocampal cultures but were most marked in area CA1. There was significant CA1 neuronal damage in cultures exposed to 10 mM glycine for 30 min or longer, or those exposed to 4 mM glycine for 24 h. The NMDA antagonists MK-801 and APV completely blocked glycine-induced neuronal degeneration in all hippocampal subfields, suggesting involvement of NMDA receptors. In contrast, a-amino-3-hydroxy-5-methyl-4-isoxazoleprionic acid (AMPA) antagonists such as CNQX, DNQX, or NBQX had no effect on glycine-induced neuronal cell death.
The problem with this in vitro model of excitotoxicity is that very high concentrations are needed to produce toxic effects of glycine. From a strictly pharmacologic point of view, it is debatable whether amounts of amino acids in the millimolar range can have some relevance to explain receptor-mediated excitotoxic mechanisms and whether the observed lesions do not simply reflect an unspecific metabolic challenge. In order to address this problem, and to extend our previous results (Newell et al. 1997), we investigated further properties of glycine-induced neurotoxicity in hippocampal slice cultures. In particular, we were interested (a) to examine the effects of glycine site NMDA agonists and antagonists on glycine toxicity, (b) to study the possible involvement of strychnine-sensitive glycine receptors, (c) to compare the toxic effects of glycine with those of glutamate in our model, and (d) to see whether combining glycine exposure with sublethal hypoxia/hypoglycemia can decrease the threshold of neurotoxicity below the millimolar range, which corresponds to the concentrations of excitatory amino acids encountered in in vivo models of cerebral ischemia.
Materials and methods
The methods of hippocampal slice preparation, exposure to glycine, and cell death assessment have been described in detail elsewhere (Newell et al. 1995a, 1995b, 1997). All experiments and surgical procedures were approved by the Animal Research Committee of the University of Washington, Seattle, Wash., USA and complied with the American legislation on animal care. Hippocampal slice cultures were prepared from 5- to 7-day-old neonatal rats (Sprague-Dawley, Bantin and Kingman Inc., Fremont, Calif., USA) according to the method of Stoppini et al. (1991) and grown on sterile transparent Anocel membranes (Whatman Inc., Clifton, N.J., USA). The growth medium consisted of 50% MEM (Gibco Laboratories, Grand Island, N.Y., USA) supplemented with hydroxyethylpiperazine ethanesulfonic acid (HEPES) and sodium bicarbonate, 25% Hank’s balanced salt solution (HBSS), 25% horse serum, and glucose to a final concentration of 6.5 mg/ml. The cultures were grown for 10–14 days before experimentation at 36.5°C, 90–100% humidity, and 5% CO. Care was taken to select healthy and morphologically intact slices prior to insult exposure.
All chemicals used in this study were purchased from Sigma Chemical Co. (St. Louis, Mo., USA). Chemicals were prepared as stock solutions in glucose-free HBSS and added as small aliquots to fresh growth medium containing the cultures to achieve the final concentrations described. Propidium iodide (PI), which rapidly enters cells with damaged membranes and becomes fluorescent after binding to nucleic acids, was added (0.5 μg/ml) to the culture medium as an indicator of neuronal cell death. The cultures were then placed in the CO2 incubator at 36.5°C for the described time intervals. Following insult exposure, the cultures were replenished with fresh growth medium with PI and returned to the incubator.
For exposure to oxygen and glucose deprivation, cultures were rinsed three times in glucose-free HBSS and then placed in HBSS without glucose. The temperature of each individual culture well was maintained at 37°C with an electronic thermometer. The cultures were then transferred into an anaerobic chamber (Forma Scientific, Marietta, Ohio, USA) pre-equilibrated to 37°C with an atmosphere of 0% oxygen, 10% hydrogen, 5% CO2, and 85% nitrogen. The hydrogen was present for interaction with a palladium catalyst that maintained the oxygen concentration at 0%. An exposure of 20 min typically did not produce visible damage to the control cultures and was defined as sublethal exposure to ischemia. Upon removal from the anaerobic chamber, the cultures were transferred to prewarmed growth medium containing PI and placed in a CO2 incubator at 36.5°C for 48 h.
For assessment of cell death, the cultures were examined using a Nikon Diaphot inverted fluorescent microscope. Fluorescent images were obtained using a Dage 72 CCD camera (Michigan City, Ind., USA) and were digitized using Optimas image analysis software (Bio-Scan Inc., Edmonds, Wash., USA). The intensity of PI fluorescence in the CA1 area was used as an index of cell death. The first measurement of fluorescent intensity was performed 40–48 h after initial exposure. The remaining neurons were killed by exposing the cultures to 3 h of anoxia. The fluorescent intensity obtained 24 h after 3 h of anoxia was set equal to 100% damage and was compared to the fluorescent intensity following the initial insult. The integrated gray values (fluorescent intensity) from CA1 following the initial insult were consequently expressed as a percentage of the value obtained following 3 h of anoxia (complete damage). The values were averaged for each group and expressed as mean±SEM. Statistical analysis of the results was performed using the Mann-Whitney test with Bonferroni correction for multiple comparisons. Significant differences were considered at P value <0.05.
Results
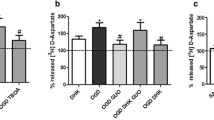
To determine if glycine site agonists of NMDA receptors were able to cause neurotoxicity, D-serine and D-alanine were administered at high doses to hippocampal cultures. In contrast to glycine’s toxic effects, exposure to 10 mM D-serine or D-alanine did not produce any fluorescence indicative of cell death in the cultures (Fig. 1a). Blockade of NMDA receptor activation by administration of the competitive glycine site antagonist 7-chlorokynurenic acid (5 mM) completely protected the hippocampal cultures against glycine toxicity (Fig. 1b). 7-chlorokynurenic acid was as effective, although less potent, as MK-801 or APV (Newell et al. 1997).

Exposure of CA1 area to glycine and the glycine site agonists D-serine and D-alanine (10 mM each) showed different effects. Whereas glycine was highly neurotoxic, D-serine and D-alanine did not produce any cell damage (*P<0.0001 compared to controls, n=12 per group). b Neurotoxicity caused by exposure to 7.5 mM glycine could be completely blocked by the competitive glycine site antagonist 7-chlorokynurenic acid (5 mM) (*P<0.0001 compared to controls, **P<0.0001 compared to glycine, n=12 per group).
Modulation of the activity of strychnine-sensitive inhibitory glycine receptors did not have any effect on glycine-induced toxicity. Agonists of glycine on its inhibitory receptor such as taurine or β-alanine did not produce any toxicity up to 10 mM (Fig. 2a). Addition of 30 μM strychnine, by itself nontoxic to hippocampal cultures, was unable to revert cell death caused by exposure to 10 mM glycine (Fig. 2b). The observation that glycine toxicity appeared to be mediated by NMDA receptors opened the possibility that the observed effects were not caused by glycine itself, but resulted from secondary synaptic release of glutamate with subsequent NMDA receptor overactivation. However, blockade of synaptic activity by addition of tetrodotoxin (1 μM) did not prevent complete cell death after exposure to high-dose glycine, suggesting that this toxic effect was not mediated by excitatory amino acid release (data not shown).

Exposure to the inhibitory glycine receptor agonists taurine or β-alanine (10 mM each) did not cause any cell damage in area CA1 (*P<0.0001 compared to controls, n=12 per group). b Addition of 30 μM strychnine, by itself nontoxic to hippocampal neurons, did not provide any protection against toxicity caused by 10 mM glycine (*P<0.0001 compared to controls, n=12 per group).
Exposure of organotypic hippocampal cultures to glutamate revealed a remarkable similarity with glycine-induced toxicity, except for the fact that glutamate caused global damage in the hippocampus without particular vulnerability of area CA1, as noted for glycine (Newell et al. 1997). Exposure to increasing doses of glutamate up to 10 mM showed that, as for glycine, cell death progressively developed between 2 and 4 mM and was complete at 8 mM (Fig. 3). In contrast to the complete blockade of glycine toxicity achieved by NMDA receptor antagonists, MK-801 (30 μM) only partially prevented neuronal damage caused by glutamate exposure, supporting the assumption that glutamate-induced toxicity is not only mediated by NMDA, but also by non-NMDA receptors (data not shown). Simultaneous exposure to increasing concentrations of glycine and glutamate in different combinations did not have any additive effect on neurotoxicity. Hippocampal cultures showed signs of cell death developing at the same concentrations (2–4 mM) for both compounds administered separately, supporting the possibility of a comparable mediation of toxicity (Fig. 4).

Comparison of the toxic effects produced by increasing doses of glutamate and glycine in area CA1. The induced damage showed a remarkable similarity between both amino acids with toxicity developing at 2–4 mM and completing at 6–8 mM. No significant difference was found between glycine and glutamate at the different concentrations tested (*P>0.05 compared to glutamate at the same concentration, n=9 per group).

Different combinations of glycine and glutamate were administered to hippocampal cultures to examine if glycine was able to potentiate glutamate-induced neurotoxicity and vice versa. a Concentrations of glycine and glutamate were simultaneously increased from 1 to 8 mM. b Glutamate was increased from 1 to 8 mM in the presence of 1 mM glycine. c Glycine was increased from 1 to 8 mM in the presence of a 1 mM glutamate. The results showed that neurotoxicity developed between 2 and 4 mM and was complete by 6–8 mM for all three protocols. No additive effect of glycine on glutamate toxicity could be demonstrated (*P>0.05 compared between groups A, B, and C for the same concentration, n=9 per group).
To further address the problem of the apparently very high concentrations needed to demonstrate glycine toxicity, we were interested to recreate pathologic conditions in our model and to observe their effects on the development of glycine-induced cell death. We reproduced pathologic ischemia-like conditions by exposing hippocampal cultures to 20 min of oxygen and glucose deprivation (Newell et al. 1995a, 1995b). Such an ischemia-like stress is not lethal in our model as no PI fluorescence can be detected in the cultures. When increasing doses of glycine or glutamate were administered in combination with sublethal oxygen/glucose deprivation, we observed that neurotoxicity developed at significantly smaller concentrations (<1 mM) than under normoxic/normoglycemic conditions (Fig. 5).

Comparison of neurotoxic effects of glycine and glutamate under sublethal ischemia-like conditions. Hippocampal cultures were exposed to sublethal oxygen/glucose deprivation (20 min) with increasing doses of glycine or glutamate. Cell death assessment showed that comparable neurotoxicity developed for glycine and glutamate at smaller concentrations than under normoxic/normoglycemic conditions (<1 mM instead of typically 2–4 mM, see Fig. 4), approaching the range of concentrations of excitatory amino acids measured during in vivo cerebral ischemia (typically 200–300 μM). This suggests that glycine can possibly contribute to ischemic neuronal injury in a similar way to glutamate (*P>0.05 compared to glutamate at the same concentration, n=9 per group).
Discussion
This study extends our previous results (Newell et al. 1997) by showing that glycine-induced neurotoxicity in hippocampal slice cultures is a selective effect of glycine not reproducible by glycine site agonists such as D-serine and D-alanine. Complete inhibition of glycine toxicity by 7-chlorokynurenic acid confirms that functioning NMDA receptors are necessary for glycine toxicity to develop. Neuronal death caused by glycine does not appear to involve strychnine-sensitive inhibitory glycine receptors. In addition, as blockade of glutamate synaptic release by tetrodotoxin did not reduce glycine toxicity, indirect participation of excitatory amino acids is probably excluded in our model. The comparability of glycine toxicity with that produced by glutamate suggests that glycine could be involved in certain neurologic disorders in which neurotoxicity mediated by excitatory amino acid plays a major role. This study also confirms that the mechanisms of NMDA receptor activation/inhibition are preserved in hippocampal slice cultures despite the very high doses required to demonstrate glycine toxicity. We assume that such supraphysiologic concentrations reflect the retained efficacy of uptake mechanisms in hippocampal cultures to remove and metabolize glycine from the synaptic cleft (Newell et al. 1997). When glycine reuptake was blocked by L-histidine, a small but distinct exacerbation of neurotoxicity was noted in our preparation (data not shown). Under sublethal ischemia-like experimental conditions, glycine- or glutamate-induced neurotoxicity developed at concentrations below 1 mM, approaching the concentrations of 200–300 μM measured during cerebral ischemia in vivo (Globus et al. 1991a; Obrenovitch and Richards, 1995). This effect probably resulted from a blockade of glycine reuptake by oxygen/glucose deprivation. It further supports the assumption that glycine may play an active role in excitotoxic neuronal injury. It also opens the possibility to study the mechanisms of glycine toxicity using lower, more physiologic amino acid concentrations than in the present study.
Glutamate NMDA receptors are ligand-gated ion channels composed of at least four transmembrane subunits (NR1, NR2A-D, NR3A-B). NR1 always has to be present to obtain a functional receptor. Different subunit compositions, posttranscriptional modifications, as well as links to intra- and extracellular signaling systems confer to the receptor multiple modulation possibilities (Dingledine et al. 1999; Cull-Candy et al. 2001). The binding site of glycine is exclusively located on the NR1 subunit, whereas the glutamate recognition site is on the NR2 subunit (Hirai et al. 1996; Foucaud et al. 2003). Glycine is necessary to enable the glutamate-activated receptor to adopt an open ion-channel configuration (Johnson and Ascher 1987; Kleckner and Dingledine 1988; Thomson 1990; Dingledine et al. 1999). Complex interactions exist between the two binding sites. Radioligand binding studies have demonstrated that glutamate and glycine reciprocally enhance the binding of the other ligand and that binding of glutamate site antagonists is enhanced by some glycine site antagonists and vice versa (Fadda et al. 1988; Monahan et al. 1990; Hood et al. 1990; Kaplita and Ferkany 1990; McBain and Mayer 1994; Sucher et al. 1996). Full activation of NMDA receptors by glycine alone seems to be possible under certain conditions (Meguro et al. 1992; Kutsawada et al. 1992; Pace et al. 1992; Chatterton et al. 2002). Pace and colleagues observed that high concentrations of neutral amino acids such as glycine and L-serine (5 mM) can elicit inward current responses in hippocampal neurons by activation of NMDA receptors. L-serine currents were completely blocked by the competitive NMDA antagonist CPP and the CPP block could be overcome by increasing concentrations of L-serine (Pace et al. 1992). In a recent study on NR3A and NR3B subunits of NMDA receptors, Chatterton et al. found that aggregations of these subunits with NR1 were able to produce functional receptors fully activated by glycine at physiologic concentrations of 3–10 μM (Chatterton et al. 2002). D-serine, usually a coagonist of conventional NMDA receptors, exerted an antagonistic effect on these NMDA receptor subtypes, particularly NR1/NR3A. Receptor activation did not request the presence of glutamate or NMDA, leading these authors to suggest that NR1/NR3A or −3B heteromeres constitute a type of excitatory glycine receptors. It is possible that overstimulation of such excitatory glycine receptors can explain some neurotoxic effects of glycine as described in the previous literature and in our present study.
Several in vitro studies have demonstrated a participation of glycine in neuronal excitotoxic injury. Glycine has been reported to markedly enhance neurotoxicity produced by NMDA in cortical cell cultures, presumably by increased activation of NMDA receptors via their glycine binding site (Shalaby et al. 1989; Patel et al. 1990; McNamara and Dingledine 1990). Interestingly, McNamara and Dingledine (1990) observed that very high concentrations of glycine (up to 3 mM) potentiated NMDA toxicity by a mechanism presumably independent of glycine binding site activation. Although glycine alone up to 1 mM was not toxic in this cortical cell culture model, their findings have some similarity with our results. First, the toxic potentiation effect of glycine necessitated concentrations 50- to 100-fold higher than those required to activate NMDA receptors. Second, selective agonists of glycine binding site such as D-serine or D-alanine were unable to potentiate NMDA toxicity, and third, glycine potentiation was competitively blocked by APV and 7-chlorokynurenic acid. Based on these findings, McNamara and Dingledine suspected an effect of strychnine-sensitive inhibitory glycine receptors. However, in the light of recent knowledge, this effect could also be ascribed to direct and full activation of certain glycine excitatory receptors by glycine alone (Chatterton et al. 2002). Glycine was shown to produce electrophysiological signs of excitotoxicity similar to those of glutamate in a model of acute hippocampal slices (Wallis et al. 1995). Extracellular application of 10 mM glycine resulted in excessive neuronal firing, followed by loss of neural transmission and slow incomplete recovery. The electrophysiological manifestations of glycine toxicity were only partially blocked by NMDA receptor antagonists. In our model using stabilized hippocampal slice cultures, the mediation of glycine toxicity by NMDA receptors was more evident and completely blocked by NMDA antagonists (Newell et al. 1997). In addition, our present results also suggest that glycine can potentiate ischemic neuronal injury in vitro.
Direct toxic effects of high extracellular glycine have been more difficult to demonstrate in vivo than in vitro (Obrenovitch and Richards 1995; Obrenovitch et al. 1997; Shoham et al. 2001). Indirect evidence is supported by the demonstration of pathologic elevations of glycine paralleling that of excitatory amino acids in experimental conditions such as cerebral ischemia (Graham et al. 1990; Globus et al. 1991a, 1991b; Baker et al. 1991; Matsumoto et al. 1993; Obrenovitch and Richards 1995; Saransaari and Oja 2001), hypoglycemia (Sandberg et al. 1986; Saransaari and Oja 2001), and head injury (Faden et al. 1989). In an animal model subjecting rats to transient global ischemia, glycine elevation persisted significantly longer than that of glutamate, suggesting the possibility of delayed toxicity in the course of stroke (Globus et al. 1991a, 1991b). Glycine is also assumed to be involved in the initiation and spread of epileptic seizures (Larson and Beitz 1988; Sierra-Paredes et al. 2001) and in the pathogenesis of schizophrenia (Waziri and Baruah 1999). Moreover, abnormal elevations of extracellular glycine have been measured in different cerebral diseases where uncontrolled release of excitatory amino acids is supposed to play an important pathophysiologic role (Kanthan and Shuaib 1995; von Essen et al. 1996; Castillo et al. 1996; Castillo et al. 1997). However, despite these observations, no clinical trial so far was able to demonstrate significant outcome improvement by reducing NMDA receptor activity with glutamate or glycine antagonists (Albers et al. 1999; Lees et al. 2000; Ikonomidou and Turski 2002; Madden 2002). In regard of the importance of excitotoxicity in human neurologic diseases, further research is warranted in this field. It is hoped that new insights in the biophysical and pharmacological properties of NMDA receptors will enable better targeting of particular subunits located in specific brain areas and open the way to new therapeutic strategies (Cull-Candy et al. 2001).
References
Albers GW, Clark WM, Atkinson RP, Madden K, Data JL, Whitehouse MJ (1999) Dose escalation study of the NMDA glycine-site antagonist licostinel in acute ischemic stroke. Stroke 30:508–513
Baker AJ, Zornow MH, Scheller MS, Yaksh TL, Stilling SR, Smullin DH, Larson AA, Kuczenski R (1991) Changes in extracellular concentrations of glutamate, aspartate, glycine, dopamine, serotonin, and dopamine metabolites after transient global ischemia in the rabbit brain. J Neurochem 57:1370–1379
Castillo J, Davalos A, Naveiro J, Noya M (1996) Neuroexcitatory amino acids and their relation in infarct size and neurological deficit in ischemic stroke. Stroke 27:1060–1065
Castillo J, Davalos A, Noya M (1997) Progression of ischaemic stroke and excitotoxic aminoacids. Lancet 349:79–83
Chatterton JE, Awobuluyl M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cul J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D (2002) Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415:793–798
Choi DW, Rothman SM (1990) The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci 13:171–182
Cull-Candy S, Brickley S, Farrant M (2001) NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11:327–335
Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51:7–61
Fadda E, Danisz W, Wroblewski JT, Costa E (1988) Glycine and D-serine increase the affinity of N-methyl-D-aspartate sensitive glutamate binding sites in rat brain synaptic membranes. Neuropharmacology 27:1183–1185
Faden AL, Demediuck P, Panter SS, Vink R (1989) The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244:798–800
Foucaud B, Laube B, Schemm R, Kreimeyer A, Goeldner M, Betz H (2003) Structural model of the N-methyl-D-aspartate receptor glycine site probed by site-directed chemical coupling. J Biol Chem 278:24011–24017
Globus MYT, Busto R, Martinez E, Valdes I, Dietrich WD, Ginsberg MD (1991a) Comparative effect of transient global ischemia on extracellular levels of glutamate, glycine, and γ-aminobutyric acid in vulnerable and nonvulnerable brain regions in the rat. J Neurochem 57:470–478
Globus MYT, Ginsberg MD, Busto R (1991b) Excitotoxic index—a biochemical marker of selective vulnerability. Neurosci Lett 127:39–42
Graham SH, Shiraishi K, Panter SS, Simon RP, Faden AI (1990) Changes in extracellular amino acid neurotransmitters produced by focal cerebral ischemia. Neurosci Lett 110:124–130
Hirai H, Kirsch J, Laube B, Betz H, Kuhse J (1996) The glycine binding site of the N-methyl-D-aspartate receptor subunit NR1: identification of novel determinants of co-agonist potentiation in the extracellular M3-M4 loop region. Proc Natl Acad Sci U S A 93:6031–6036
Hood WF, Compton RP, Monahan JB (1990) N-methyl-D-aspartate recognition site ligands modulate activity at the coupled glycine recognition site. J Neurochem 54:1040–1046
Ikonomidou C, Turski L (2002) Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 1:383–386
Johnson JW, Ascher P (1987) Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325:529–531
Kanthan R, Shuaib A (1995) Clinical evaluation of extracellular amino acids in severe head trauma by intracerebral in vivo microdialysis. J Neurol Neurosurg Psychiatry 59:326–327
Kaplita PV, Ferkany JW (1990) Evidence for direct interactions between the NMDA and glycine recognition sites in brain. Eur J Pharmacol 188:175–179
Kleckner NW, Dingledine R (1988) Requirement for glycine in activation of NMDA receptors expressed in Xenopus oocytes. Science 241:835–837
Kutsawada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meduro H, Masaki H, Kumanishi T, Arakawa M, Mishina M (1992) Molecular diversity of the NMDA receptor channel. Nature 358:36–41
Larson AA, Beitz AJ (1988) Glycine potentiates strychnine-induced convulsions: role of NMDA receptors. J Neurosci 8:3822–3826
Lees KR, Asplund K, Carolei A, Davis SM, Diener HC, Kaste M, Orgogozo JM, Whitehead J (2000) Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: a randomised controlled trial. GAIN International Investigators. Lancet 255:1949–1954
Lipton SA, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613–622
Madden K (2002) NMDA receptor antagonists and glycine site NMDA antagonists. Curr Med Res Opin 18 [Suppl 2]:27–31
Matsumoto K, Graf R, Rosner G, Taguchi J, Heiss WD (1993) Elevation of neuroactive substances in the cortex of cats during prolonged focal ischemia. J Cereb Blood Flow Metab 13:586–594
McBain CJ, Mayer ML (1994) N-methyl-D-aspartatic acid receptor structure and function. Physiol Rev 74:723–760
McNamara D, Dingledine R (1990) Dual effect of glycine on NMDA-induced neurotoxicity in rat cortical cultures. J Neurosci 10:3970–3976
Meguro H, Mori H, Araki K, Kushiya E, Kutsuwada T, Yamazaki M, Kumanishi T, Arakawa M, Sakimura K, Mishina M (1992) Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature 357:70–74
Monahan JB, Biesterfeldt JP, Hood WF, Compton RP, Cordi AA, Vazquez MI, Lanthorn TH, Wood PL (1990) Differential modulation of the associated glycine recognition site by competitive N-methyl-D-aspartate receptor antagonists. Mol Pharmacol 37:780–784
Newell DW, Barth A, Papermaster V, Malouf AT (1995a) Glutamate and non-glutamate receptor mediated toxicity caused by oxygen and glucose deprivation in organotypic hippocampal cultures. J Neurosci 15:7702–7711
Newell DW, Barth A, Malouf AT (1995b) Glycine site NMDA receptor antagonists provide protection against ischemia-induced neuronal damage in hippocampal slice cultures. Brain Res 675:38–44
Newell DW, Barth A, Ricciardi TN, Malouf AT (1997) Glycine causes increased excitability and neurotoxicity by activation of NMDA receptors in the hippocampus. Exp Neurol 145:235–244
Obrenovitch TP, Richards DA (1995) Extracellular neurotransmitter changes in cerebral ischemia. Cerebrovasc Brain Metab Rev 7:1–54
Obrenovitch TP, Hardy AM, Urenjak J (1997) High extracellular glycine does not potentiate N-methyl-D-aspartate-evoked depolarization in vivo. Brain Res 746:190–194
Pace JR, Martin BM, Paul SM, Rogawski MA (1992) High concentrations of neutral amino acids activate NMDA receptor currents in rat hippocampal neurons. Neurosci Lett 141:97–100
Patel J, Zinkand WC, Thompson C, Keith R, Salama A (1990) Role of glycine in the N-methyl-D-aspartate-mediated neuronal cytotoxicity. J Neurochem 54:849–854
Perry TL, Hansen S (1981) Amino acid abnormalities in epileptogenic foci. Neurology 31:872–876
Sandberg M, Butcher SP, Hagberg H (1986) Extracellular overflow of neuroactive amino acids during severe insulin-induced hypoglycemia: in vivo dialysis of the rat hippocampus. J Neurochem 47:178–184
Saransaari P, Oja SS (2001) Characteristics of hippocampal glycine release in cell-damaging conditions in the adult and developing mouse. Neurochem Res 26:845–852
Shalaby I, Chenard B, Prochniak M (1989) Glycine reverses 7-chlorokynurenate blockade of glutamate neurotoxicity in cell culture. Eur J Pharmacol 160:309–311
Shoham S, Javitt DC, Heresco-Levy U (2001) Chronic high-dose glycine nutrition: effects on rat brain cell morphology. Biol Psychiatry 49:876–885
Sierra-Paredes G, Senra-Vidal A, Sierra-Marcuno G (2001) Effect of extracellular long-time microperfusion of high concentrations of glutamate and glycine on picrotoxin seizure thresholds in the hippocampus of freely moving rats. Brain Res 888:19–25
Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182
Sucher NJ, Awobuluyi M, Choi YB, Lipton SA (1996) NMDA receptors: from genes to channels. Trends Pharmacol Sci 17:348–355
Thomson AM (1990) Glycine is a coagonist at the NMDA receptor/channel complex. Prog Neurobiol 35:53–74
von Essen C, Rydenhag B, Nyström B, Mozzi R, van Gelder N, Hamberger A (1996) High levels of glycine and serine as a cause of the seizure symptoms of cavernous angiomas? J Neurochem 67:260–264
Wallis RA, Panizzon KL, Nolan JP (1995) Glycine-induced CA1 excitotoxicity in the rat hippocampal slice. Brain Res 685:225–235
Waziri R, Baruah S (1999) A hyperglycinergic rat model for the pathogenesis of schizophrenia: preliminary findings. Schizophr Res 37:205–215
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Barth, A., Nguyen, L.B., Barth, L. et al. Glycine-induced neurotoxicity in organotypic hippocampal slice cultures. Exp Brain Res 161, 351–357 (2005). https://doi.org/10.1007/s00221-004-2079-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-004-2079-7