
Abstract
SARS-CoV-2, the causative agent of COVID-19, continues to cause global morbidity and mortality despite the increasing availability of vaccines. Alongside vaccines, antivirals are urgently needed to combat SARS-CoV-2 infection and spread, particularly in resource-limited regions which lack access to existing therapeutics. Small molecules isolated from medicinal plants may be able to block cellular entry by SARS-CoV-2 by antagonising the interaction of the viral spike glycoprotein receptor-binding domain (RBD) with the host angiotensin-converting enzyme II (ACE2) receptor. As the medicinal plant Gunnera perpensa L. is being used by some South African traditional healers for SARS-CoV-2/COVID-19 management, we hypothesised that it may contain chemical constituents that inhibit the RBD-ACE2 interaction. Using a previously described AlphaScreen-based protein interaction assay, we show here that the DCM:MeOH extract of G. perpensa readily disrupts RBD (USA-WA1/2020)-ACE2 interactions with a half-maximal inhibition concentration (IC50) of < 0.001 µg/mL, compared to an IC50 of 0.025 µg/mL for the control neutralising antibody REGN10987. Employing hyphenated analytical techniques like UPLC-IMS-HRMS (method developed and validated as per the International Conference on Harmonization guidelines), we identified two ellagitannins, punicalin (2.12% w/w) and punicalagin (1.51% w/w), as plant constituents in the DCM:MeOH extract of G. perpensa which antagonised RBD-ACE2 binding with respective IC50s of 9 and 29 nM. This good potency makes both compounds promising leads for development of future entry-based SARS-CoV-2 antivirals. The results also highlight the advantages of combining reverse pharmacology (based on medicinal plant use) with hyphenated analytical techniques to expedite identification of urgently needed antivirals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of the coronavirus disease 2019 (COVID-19), was first reported in late 2019, in Wuhan, China, and has since become an unprecedented and persistent threat to global health and economies [1,2,3]. While multiple anti-SARS-CoV-2 vaccines are currently available, emergence of new SARS-CoV-2 variants is already undermining their efficacy [4]. Additionally, while newly developed therapeutic antibodies and antivirals may offer new options for COVID-19 treatment following SARS-CoV-2 infection, their global use is likely to be hampered, at least in the near term, due to limited access and/or insufficient in-patient resources in many low- and middle-income countries. As a result, discovery and development of additional SARS-CoV-2 antivirals remain an urgent priority.
The genome of SARS-CoV-2, a positive-sense single-stranded RNA virus, encodes for structural proteins including the spike glycoprotein, membrane, nucleocapsid, envelope and other accessory proteins which are required for the viral replication cycle and represent attractive antiviral targets [5]. For example, remdesivir, currently one of the only drugs to gain FDA approval for COVID-19 despite limited efficacy, is a broad-spectrum antiviral agent which targets the RNA-dependent RNA polymerase enzyme [6]. More recently, molnupiravir and Paxlovid (PF-07321332 + ritonavir) have been developed to inhibit SARS-CoV-2 replication by inhibiting the RNA-dependent RNA polymerase or SARS-CoV-2 main protease (3CL protease), respectively [7, 8]. Another promising therapeutic target is the binding and entry of SARS-CoV-2 which is regulated by the viral spike glycoprotein [9]. In particular, the receptor-binding domain (RBD) of the spike S1 subunit interacts with the host angiotensin-converting enzyme 2 (ACE2) receptor to promote SARS-CoV-2 binding [10]. With additional segments of the spike protein assisting in membrane fusion, the SARS-CoV-2 virus gains entry into the host cell upon cleavage by furin, TMPRSS2, and other host proteases [11]. As a result, compounds that can disrupt the spike RBD-ACE2 interaction would also be expected to inhibit SARS-CoV-2 entry and replication. However, no small molecule antivirals are currently approved to block SARS-CoV-2 entry, while current therapeutic antibody treatments are limited in much of the world due to cost, stability, and the need for in-patient infusion resources.
Pure compounds isolated from natural products routinely serve as leads for the development of new pharmaceuticals [12,13,14] including as potential agents against SARS-CoV-2 [15,16,17,18]. The medicinal plant Gunnera perpensa L., a member of the Gunneraceae family, is widely distributed throughout tropical Africa and frequently used by traditional healers for its extensive medicinal properties [19]. For example, decoctions of the leaves, stems, rhizomes, and roots are used to treat numerous diseases and health problems including infertility, colic, stomach ailments, and colds [20]. Most recently, the dried plant powder has been used by traditional healers of the Eastern Cape, South Africa, to treat suspected COVID-19 exposure or suspected COVID-19 cases (F.J. Isaacs, personal communication).
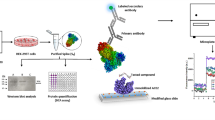
Based on this information, we hypothesised that components of G. perpensa L. may exhibit potent in vitro bioactivities consistent with targeting SARS-CoV-2. However, the isolation of bioactive compounds from complex natural product mixtures constitutes a major limitation of this strategy, particularly given the urgency to develop a treatment during an ongoing pandemic such as COVID-19. Toward addressing this limitation, advanced hyphenated analytical techniques including ultra-performance liquid chromatography, ion mobility separation, and high-resolution mass spectrometry (UPLC-IMS-HRMS) were used to expedite the identification of two active compounds from G. perpensa, punicalin and punicalagin (Fig. 1), and characterise their ability to inhibit the interaction of the spike glycoprotein of the SARS-CoV-2 with the ACE2 using an established AlphaScreen-based protein interaction assay [18]. Results highlight the importance of combining several optimised analytical techniques with leads obtained from reverse pharmacology approaches to ensure the rapid isolation and identification of bioactive compounds and potential SARS-CoV-2 antiviral leads from complex natural product mixtures.

Graphical representation of the bioassay-guided fractionation and eventual identification of punicalin and punicalagin from the G. perpensa crude extraction
Materials and methods
Spike protein and reagents
The pure standards, punicalagin and punicalin, were purchased from Merck, South Africa (Merck, Darmstadt, Germany). Analytical grade (AR) extraction solvents, i.e. dichloromethane (DCM), methanol (MeOH) and formic acid (FA), were purchased from Merck, South Africa. The HPLC grade (super purity) solvents acetonitrile (ACN) and methanol used in the fractionation and purification stages were purchased from Romil-SpS™, Microsep, South Africa (Waterbeach, Cambridge, UK), and the ethyl acetate (EtOAc) from Sigma-Aldrich (Merck, Darmstadt, Germany). ACE2-Fc and HIS-tagged SARS-CoV-2 Spike-RBD (USA-WA1/2020 variant) proteins were purchased from Sino Biological (Chesterbrook, PA, USA). REGN10987 was obtained from excess aliquot volumes at the Perelman School of Medicine at the University of Pennsylvania which could not be used for patients (a gift from Dr. Pablo Tebas).
Preparation of the G. perpensa DCM:MeOH extract
Arial parts of G. perpensa were collected from a cultivation site in Uitenhage, Eastern Cape, South Africa. A voucher specimen (PRU 128787) was prepared and deposited at the University of Pretoria’s H.G.W.J. Schweickerdt Herbarium. The leaves were air-dried and ground into a fine powder using a hammer mill. Powdered leaves (12 g) were loaded into an in-house custom-made glass extraction vessel, fitted with a teflon stopcock and a sintered glass filter (porosity 3). A 50-mL 1:1 mixture of DCM:MeOH was added to the vessel containing the plant material and placed in an ultrasonic bath (power—005, frequency—high, time—1 h). Thereafter, the DCM:MeOH solution was drained before a subsequent extraction cycle with 50 mL MeOH. The two separate extract solutions were combined and dried to completion using an SP Genevac HT6 (Genevac Ltd., Ipswich, UK).
Primary fractionation of the DCM:MeOH extract
The resulting dry extract was fractionated as previously described by Thornburg et al. [21]. A HypeSep C8 SPE (solid phase extraction) cartridge (2 g/6 mL) was used to fractionate the extract into 7 fractions using a Gilson GX-241 ASPEC® liquid handler fitted with a Gilson Verity® 4060 pump controlled with TRILUTION® software. The DCM:MeOH extract (250 mg) was dissolved in 5 mL of a MeOH:EtOAc (3:2) solution and adsorbed onto a cottonwool roll. The cottonwool with the extract was dried in a SP Genevac HT6 before being transferred to an empty 10-mL SPE cartridge. Prior to fractionation, the C8 SPE cartridge was prepared by washing it with 3 column volumes of 100% MeOH before conditioning it with 3 column volumes of the first eluent, viz. 19:1 (H2O:MeOH). Fractionation followed by connecting the loaded SPE cartridge containing the cottonwool with the adsorbed extract in series above the conditioned C8 cartridge and injecting 1 column volume of each of the following eluent systems via positive pressure into the cottonwool loaded SPE cartridge: 19:1 (H2O:MeOH); 4:1 (H2O:MeOH); 3:2 (H2O:MeOH); 1:1 (H2O:MeOH); 2:3 (H2O:MeOH); 1:4 (H2O:MeOH); and 1:1 (ACN:MeOH). The fractions were collected separately and dried in a SP Genevac HT6 prior to submission for bioscreening analysis.
AlphaScreen binding assays
AlphaScreen assays were performed as described previously [18]. Briefly, 2 nM of ACE2-Fc was incubated with 5 nM HIS-tagged SARS-CoV-2 spike RBD in the presence of 5 μg/mL nickel chelate donor beads in a total of 10 μL of 20 mM Tris (pH 7.4), 150 mM KCl, and 0.05% CHAPS. Test samples were diluted to 100 × final concentration in dimethyl sulfoxide (DMSO). 5 μL of ACE2-Fc/protein A acceptor bead was then added to the reaction, followed by 100 nL test compound and 5 μL of RBD-HIS/nickel chelate donor beads. All conditions were performed in duplicate. Following incubation at room temperature for 2 h, luminescence signals were measured using a ClarioStar plate reader (BMC Labtech, Cary, NC, USA). Data were normalised to percent inhibition, where 100% equalled the AlphaScreen signal in the absence of RBD-HIS, and 0% denoted signal in the presence of both protein and DMSO vehicle control.
Half-maximal inhibitory concentrations (IC50) were determined using GraphPad Prism v. 9.1.2 (GraphPad Software, San Diego, CA, USA). Data from extracts and REGN109987 are presented as the mean ± S.D. from at least 2 independent experiments. Data from pure compounds and antibodies are presented as the mean ± S.D. from 3 independent experiments.
UPLC-IMS-HRMS chromatographic conditions
Due to the complex nature of the fractions following SPE, UPLC-IMS-HRMS was employed to analyse the primary and tertiary fractions together with the crude extract in order to accurately acquire the monoisotopic mass, retention time, and ion mobility data. UPLC analysis was performed using a Waters Acquity UPLC system (Waters Corp., MA, USA), which is equipped with both an auto sampler and a binary solvent delivery system. The DCM:MeOH extract and fractions were prepared by dissolving them in a H2O:ACN (4:1) solution before filtering them through a 0.22-μm syringe filter. The samples were analysed on a ACQUITY UPLC® HSS T3 (2.1 × 150 mm, 1.8 µm) column (Waters Inc., Milford, MA, USA), which showed the best selectivity for the compounds. A gradient method separation was employed using H2O (0.1% FA, 0.01% ammonium formate) and ACN (0.1% FA) as the two solvents. The method conditions were as follows: 97% H2O (0.1% FA), held for 0.1 min, followed by a linear gradient increase to 100% ACN at 14 min. A 2-min washing hold was used (14–16 min) before reconditioning the column with the starting conditions (16.5–20 min). To ensure repeatable results, the column temperature was held constant at 40 °C with a set flow rate of 0.4 mL/min and an injection volume of 5 μL.
UPLC-IMS-HRMS instrumentation and MS conditions
A Waters® Synap G2 high-definition mass spectrometer (HDMS) system (Waters Inc., Milford, MA, USA) was used for compound separation and detection. The system is setup with a Waters ACQUITY UPLC® hyphenated to a quadrupole mass filter. Additionally, the system comprises of a Triwave™ ion mobility (IM) cell and a high-resolution time-of-flight (TOF) mass analyser. MassLynx™ v. 4.1 (Waters Inc., Milford, MA, USA) software was used to operate the instrument and for data acquisition. Sodium formate clusters were used to calibrate the MS using the Intellistart software function over a mass range of 50–1200 Da. The MS source parameters were optimised for ESI negative mode and were set as follows: source temperature of 120 °C, extraction cone voltage of 4.0 V, sampling cone of 25.0 V, cone gas flow of 10 L/h, desolvation temperature of 350 °C, desolvation gas flow of 600.0 L/h, and a capillary voltage of 2.4 kV for the negative mode. An internal lockmass control standard is used, a 2 ng/µL solution of leucine enkephalin (m/z 555.2693). The lockmass standard is infused directly into the source, added at a rate of 3 µL/min to account for experimental drift in mass by a secondary orthogonal electrospray ionisation probe. Infusion is done intermittently every 10 s.
UPLC-IMS-HRMS IMS conditions
For the ion mobility spectrometry (IMS), nitrogen gas was used at a flow rate of 90 mL/min and a helium gas flow of 180 mL/min was set for the helium cell. The IMS wave velocity was set at 650 m/s with the wave height set to 40.0 V and wave velocity ramp of 300 to 600 m/s. A transfer wave velocity of 191 m/s was used with a transfer wave height of 0.1 V. The trap DC bias was set at 45.0 V with the helium cell DC voltage set at 35.0 V. Additionally, a mobility separation delay of 450 µs was used. The IMS was calibrated prior to sample analysis using the Waters Major Mix Calibration Sample with Driftscope (v. 2.8). This enabled the determination of the experimental collisional cross section (CCS) values (Ω). The CCS error was determined as < 3.7% (0.66 ± 0.8%).
Data acquisition in UPLC-IMS-HRMS analysis
Mass spectral scans were collected every 0.3 s. Raw data was collected in a continuous form, with mass to charge ratios (m/z) of 50 to 1200 Da recorded. Data was collected in data-independent acquisition (DIA) mode using two functions with a low and high collision energy (MSE). The collision energies were maintained at 10 V for the low MS transfer collision energy and 30 V for high MS transfer collision energy.
UPLC-IMS-HRMS data processing
All data processing was performed using UNIFI® Scientific Information System (Waters Inc., Milford, MA, USA). To ensure optimum processing, the following settings were used: automatic chromatographic peak width settings for the run entirety and automatic intensity threshold. Additionally, the high and low energy functions had an intensity threshold set to 5.0 and 5.0 detector counts respectively. A tolerance window for the retention time was set to ± 0.1 min preventing artefact peaks and those that were too wide. A target mass tolerance of 15.0 ppm was set with a fragmentation match tolerance of ± 10.0 mDa with a maximum relative intensity threshold of 0.01 for targets with the same m/z ratio. A 5.0% CCS tolerance was set. Lock mass correction of all data was done with UNIFI® automatically prior to data processing.
G. perpensa secondary and tertiary fractionation
Based on the data generated from the biological screens, targeting the RBD/ACE2 interaction was prioritised for further work. Two compounds, punicalin and punicalagin, were tentatively identified in primary fraction 3 using UPLC-IMS-HRMS, and hypothesised to be the active constituents contributing to the RBD/ACE2 activity based on prior published literature of the phenolic class of compounds [15]. To target these compounds contributing to the selective inhibition of RBD/ACE2 binding, the most potent primary fraction in the RBD/ACE2 screens, primary fraction 3, was further purified on a Buchi Pure C-815 Flash system (Buchi, Flawil, Switzerland) fitted with a UV detector and an ELSD. A Buchi EcoFlex C18 (50 µm, 20 g) cartridge was used, and a gradient elution method employed. The solvent system used consisted of solvent A and solvent B, namely H2O (0.1% FA) and MeOH (0.1% FA), respectively. The column was equilibrated for 6 min with the starting conditions before the following elution method was used at a flow rate of 40 mL/min: an initial hold at 5% MeOH for 0.5 min before a linear increase to 8% MeOH over 1.3 min, a second linear increase of solvent B to 21% MeOH over 8.2 min, and a final linear increase to 100% MeOH over 4.4 min. A 25-min hold followed to ensure adequate washing of the column. Wavelengths (λ) 254 nm, 265 nm, 230 nm, and 320 nm were monitored, and collection automatically triggered where an excess absorbance of 0.05 AU was detected.
Following HPLC–UV/MS of the 3 secondary fractions produced from the Buchi flash system, punicalin and punicalagin were tentatively identified in secondary fraction 1 and secondary fraction 2 based on their m/z and observed maximum absorbances (λmax) at 217 nm, 258 nm, and 378 nm, comparable to published data [22]. Secondary fractions 1 (230 mg) and 2 (150 mg) were combined and underwent a final purification step, with the use of preparative high-performance liquid chromatography (prep HPLC). A Waters chromatographic system was used, equipped with a Waters photodiode array (PDA) detector (Model 2998) and ACQUITY QDa detector (Waters, Milford, MA, USA). The combination of secondary fractions 1 and 2 was prepared by dissolving it in a H2O:MeOH (4:1) solution before filtering the sample with a 0.22-μm syringe filter. The sample was further purified on the HPLC by performing a single injection, under ES negative MS conditions, on an Xbridge® Prep C18 OBD™ (19 × 250 mm, 5 μm) column. The analytical gradient method used to screen the secondary fractions was modified for a preparatory column. The method used consisted of H2O (0.1% FA) as solvent A and MeOH (0.1% FA) as solvent B at a flow rate of 19.0 mL/min. The chromatographic method used was as follows: 5% MeOH for 5 min, a linear gradient increase to 100% MeOH (5–30 min), a 4-min washing hold (30–34 min) followed by linear return to starting conditions (34–43 min). A timed collection approach was followed in which collection was triggered every 1.5 min. Qda settings were optimised as follows: ES negative mode with a mass range of 150 to 1100 Da. The probe temperature was set to 500 °C with the capillary voltage set to 0.81 kV, cone voltage of 13.75 V, and a source temperature of 120 °C.
Tertiary fractions generated using mass-directed fractionation on the prep HPLC were rescreened in RBD/ACE2 interaction assays as described in the ‘AlphaScreen binding assays’ section. Tertiary fraction 8 (22.5 mg), amongst others, possessed potent antagonistic activity against RBD/ACE2 interaction and was further analysed on UPLC-IMS-HRMS.
Conclusive identification of compounds in the bioactive tertiary fractions generated from preparative HPLC
Tertiary fractions presenting RBD/ACE2 antagonistic behaviour were analysed, together with the crude and bioactive primary fractions on a UPLC-IMS-HRMS. Monoisotopic mass, MS/MS fragmentation patterns, molecular formulae, and % error values corresponded to those of punicalagin and punicalin. To conclusively confirm this, punicalin and punicalagin standards were purchased and analysed on UPLC-IMS-HRMS together with the bioactive extract and fractions. Stock standard solutions of the 2 ellagitannins, punicalin and punicalagin, were prepared by dissolving the compounds in a 4:1 (H2O:ACN) before further dilution to yield working stock solutions of 200 ppm. The MS/MS fragmentation pattern, accurate monoisotopic mass, retention time, CCS (collisional cross section) data, and drift time were used to confirm the presence of the compounds in the bioactive extract and fractions. The compounds punicalagin and punicalin were reported as ‘found’ if their retention time (RT) values were within 0.1 min of the peaks in the purchased standards, and their CCS values were within 5% of that of the purchased standards. Additionally, the purchased standards were rescreened in RBD/ACE2 inhibition assays to further confirm that the compounds were responsible for the bioactivity in the plant.
Isolation and characterisation of punicalagin
Tertiary fraction 8, produced by HPLC–UV/MS, was further purified on an Agilent 1260 Infinity Series HPLC (Agilent Technologies, Santa Clara, CA, USA) with an online SPE solution in combination with a Bruker amazon SL IonTrap mass spectrometer (Bruker Daltonics, Bremen, Germany). A 13 mg/mL solution was prepared and a Luna® C18(2) (250 × 4.6 mm, 5 μm) column was used for separation together with Oasis® On-Line SPE trapping cartridges for collection. The following gradient method was used with H2O (0.1% FA) and MeOH (0.1% FA) as the mobile phases with a 12-µL injection volume and a flow rate of 0.5 mL/min: 20% MeOH isocratic hold for 15 min, linear increase to 100% MeOH at 15.5 min, an isocratic hold at 100% MeOH until 20 min, before returning to starting conditions at 23 min. A 5-min column equilibration phase with the starting conditions was incorporated before the subsequent injection.
Mass-directed fractionation was used in ESI negative mode and peaks corresponding to 541 g/mol (λmax = 217 nm, 258 nm, and 378 nm) were trapped.
Punicalagin was isolated and analysed on a 400 MHz Bruker Advance III NMR spectrometer at 25 °C. The compound was dissolved and analysed in deuterated acetone-d6 (Sigma-Aldrich, Milwaukee, WI, USA). The resulting spectrum was compared to that published in literature [23].
UPLC-IMS-HRMS method validation
The UPLC-IMS-HRMS method used for G. perpensa was partially validated according to the International Conference on Harmonization (ICH) guidelines [24] for specificity, linearity, precision, and quantification parameters, namely limit of detection (LOD) and limit of quantification (LOQ).
Specificity
To ascertain the specificity of the UPLC-IMS-HRMS method used, punicalin and punicalagin standards together with the blanks and DCM:MeOH extract were analysed on the same UPLC-IMS-HRMS instrument, employing the same chromatographic conditions and injection volumes, on the same day. Comparison of the spectral peaks and retention times was used to ascertain the selectivity of the analytical method used.
Linearity
To validate the linearity of the UPLC-IMS-HRMS method employed, standard solutions of the ellagitannins were prepared in the range of 1–50 μg/mL and 5–80 μg/mL for punicalin and punicalagin, respectively. Each sample was analysed in triplicate. A 5-point calibration curve was constructed by plotting the peak areas versus the respective analyte concentrations using linear regression analysis.
Precision
The method precision was evaluated on a repeatability (precision under the same conditions within a short period of time, i.e. intra-day) level. The intra-day variations of the analytical method were determined by analysing the independently prepared samples on the same day.
Limit of detection (LOD) and limit of quantification (LOQ)
The LOD and LOQ were calculated with the use of the following Eqs. (1) and (2), respectively:
and
where σ and S denote the standard deviations of the response and the gradient of the calibration curve, respectively.
Quantification of the ellagitannins
Quantification of the compounds of interest, viz. punicalin and punicalagin, was carried out with the use of a 5-point calibration curve, prepared for punicalin and punicalagin respectively. These standard solutions were prepared by serial dilution of the original working solutions and used for subsequent quantification of the compounds in the DCM:MeOH extract. The concentrations of the 2 compounds were determined on a dry weight basis (%w/w) of the DCM:MeOH extract. The total punicalin and punicalagin concentrations were reported.
Results
Extraction, fractionation using SPE, UPLC-MS analysis, and inhibition of RBD-ACE2 interaction
To isolate the bioactive compounds, a bioassay guided fractionation approach was adopted in this study (Fig. 1). From 12 g of air-dried G. perpensa leaves, 1.1 g of dried crude extract was obtained (9.2% yield) after a 2-step extraction procedure with dichloromethane and methanol (DCM:MeOH). A 250-mg portion of the crude DCM:MeOH extract was subjected to fractionation using a C8 SPE cartridge, yielding 7 primary fractions of varying mass, ranging between 10.5 and 24 mg (Table 1). The fractions ranged from highly polar 19:1 (H2O:MeOH) to moderately polar 1:1 (ACN:MeOH) in nature.
To determine whether the G. perpensa DCM:MeOH extract and/or the primary fractions may contain components that disrupt SARS-CoV-2 cellular entry, we made use of a previously described AlphaScreen technology-based assay [18] which uses a SARS-CoV-2 RBD protein (USA-WA1/2020 sequence) containing a C-terminal HIS tag, bound to a nickel chelate acceptor bead, in addition to a full-length ACE2 peptide with a C-terminal Fc tag bound to a donor protein A bead. When an RBD-ACE2 binding event occurs, the two beads are brought into proximity of each other such that excitation at 680 nm results in a singlet oxygen transfer and luminescence at 615 nm. Using this assay, we showed that the clinically approved neutralising antibody REGN10987 (imdevimab), which targets the RBD-ACE2 interaction [25], could inhibit luminescence due to RBD-ACE2 binding with dose dependence and with an IC50 of 0.025 μg/mL (Fig. 2; Table S1). Of the samples screened, the DCM:MeOH extract and primary fractions 2–7 also demonstrated potent activity in the AlphaScreen assay, with IC50s in the low ng/mL range (Fig. 2; Table S1). Primary fraction 3 was of particular interest, based on the relatively high yield (Table 1) and activity observed in the AlphaScreen assay (IC50 < 0.001 μg/mL).

Dose–response curves denoting ability of G. perpensa DCM:MeOH extract, primary fractions, and control neutralising antibody REGN10987 to disrupt luminescence due to SARS-CoV-2 spike RBD-ACE2 protein binding in AlphaScreen assay
Based on the potent activity of primary fraction 3 in the RBD-ACE2 interaction assay (Fig. 2), the crude extract and primary fraction were analysed by UPLC-IMS-HRMS. Fifteen prominent peaks were observed in the primary fraction, of which only 2 were identified. These two were also present in the extract, providing confirmation that they were not artefacts introduced during the purification process. The accurate mass, formulae, and fragments of 2 minor peaks common to primary fraction 3 and the extract matched those of 2 known ellagitannins, punicalin and punicalagin (Fig. 3), of which punicalagin was previously reported to occur within G. perpensa [19]. Based on previous literature claiming potent antiviral activity of punicalagin, it was hypothesised to be the active constituent [15]. Due to the complexity of the primary fraction, further purification steps were undertaken to isolate the ellagitannins.

The chemical structures of punicalin (a) and punicalagin (b)
G. perpensa secondary and tertiary fractionations and evaluation of RBD-ACE2 interaction
To further assist in active compound/s identification from primary fraction 3, 520 mg of the fraction was fractionated on a Buchi Pure C-815 Flash system, yielding 3 secondary fractions. Secondary fraction 1 (230 mg) and secondary fraction 2 (150 mg) were combined based on their HPLC–UV/MS profile which showed the presence of punicalagin based on the observed m/z 541 [M-2H]2− and observed maximum absorbances (λmax) at 217 nm, 258 nm, and 378 nm, which compared well to published data [22]. The combined secondary fractions were further purified on a preparatory HPLC–UV/MS instrument, yielding 13 tertiary fractions, named tertiary fractions 1–13. These tertiary fractions were evaluated in RBD-ACE2 bioassays to identify the active fractions to assist in identifying the active compounds contained in them. All fractions were bioassayed to ensure that no artefacts were formed during the chromatography steps that could contribute to the bioactivity. Of the samples screened in the AlphaScreen assays, most secondary fractions displayed good activity, with IC50s in the low μg/ml range (Fig. 4). In particular, several fractions demonstrated exceptional activity in the AlphaScreens, with IC50s in the low ng/mL range (Fig. 4).

Dose–response curves denoting ability of tertiary subfractions to disrupt luminescence due to SARS-CoV-2 spike RBD—host ACE2 protein binding in AlphaScreen assay
Chemical profiling and identification of the active constituents
Based on the antagonistic activity of tertiary fractions 2–8 in RBD-ACE2 interaction assays (Fig. 4), the crude DCM:MeOH extract (Fig. 5) and fractions (Fig. 6) were analysed on a UPLC-IMS-HRMS to identify the active compounds. UPLC-IMS-HRMS analysis, in ESI negative mode, showed the presence of 3 main peaks with m/z ratios of 781.0586 and 541.0251 (Figs. 5 and 6). Their monoisotopic mass and molecular formulae corresponded to the [M-H]− quasi-molecular ion of punicalin (C34H22O22) and the [M-2H]2− double charged quasi-molecular ion of punicalagin (C48H28O30) with a mass error of 2.0 ppm and − 4.8 ppm observed, respectively.

BPI chromatogram of the bioactive G. perpensa DCM:MeOH crude extract in ESI negative mode

Partial BPI chromatogram of the bioactive tertiary fraction 3 in ESI negative mode. Three major peaks (annotation highlighted) are hypothesised to be that of punicalin (m/z 781.0546) (peak 1) and α/β-punicalagin (m/z 541.0251) (peak 2/3) as a double charged quasi-molecular ion
To unequivocally confirm the identity of the 2 compounds observed in the bioactive tertiary fractions, subsequent confirmation was also carried out with the use of commercially available standards purchased from Merck. The standards were prepared and analysed concurrently with the bioactive fractions and crude extracts in both ESI negative mode and ESI positive mode on a UPLC-IMS-HRMS. However, due to the chemical nature of the compounds, the ellagitannins were found to exclusively ionise in ESI negative mode. The monoisotopic mass, MS/MS fragmentation pattern, molecular formula, drift time, and CCS values of those observed in the sample were compared to those found for the standards. Punicalin (peak 1, Fig. 6) appeared at a m/z 781.0586 [M-H]− with a molecular formula of C34H22O22 and a mass error of 2.0 ppm. The observed fragments (Table 2) matched those of the standard and other published data [26]. The two separate peaks observed in the tertiary fraction (peak 2/3, Fig. 6) corresponded to the anomers of punicalagin. Punicalagin exists as a reversable mixture of α/β anomers [27]. The α/β-punicalagin appeared as doubly charged quasi-molecular ion at a m/z 541.0251 with a molecular formula of C48H28O30 and an observed mass error of − 4.8 ppm. The observed fragments of punicalin and punicalagin in the DCM:MeOH extract also matched those in literature and those of the purchased standard. The m/z 301 observed corresponds to ellagic acid, a moiety of punicalagin (Fig. 7) [26]. Additionally, drift time and CCS values observed corresponded to those observed for the purchased standard (Table 2). Although no significant differences in CCS nor drift time were observed for the same quasi-molecular ions of the separate anomers, expectedly, clear differences in CCS and drift time are observed for the different quasi-molecular ions of the same anomer (Table 3), providing an interesting 3rd dimension of separation (Fig. 8).

Punicalagin [M-2H]2− with its (a) low and (b) high energy mass spectra with the accompanying fragments detected and identified in the G. perpensa L. crude extract

An ion mobility contour plot of the punicalagin standard, generated by UNIFI® with retention time (min) versus drift time (ms) providing a 3rd dimension of separation, clearly separating the different quasi-molecular ions in the same peak (left). An ion mobility contour plot of the punicalagin standard, with drift time (ms) versus m/z (Da) showing different drift times for the 2 separate quasi-molecular ions of the respective α/β peaks (right)
Punicalagin (1.0 mg) was isolated in the form of a yellow amorphous powder from tertiary fraction 8 (22.50 mg) on an Agilent LC-SPE and characterised on a 400 MHz Bruker Advance III NMR spectrometer (Fig. S1). The chemical shifts were compared to those described in literature for the two anomers [23] and compared favourably to punicalagin. Additionally, the α-punicalagin anomeric hydrogen was detected on the 1H-NMR as a doublet at 5.09 ppm (1H, d, J = 3.67 Hz) and a doublet at 4.76 ppm (1H, d, J = 8.56 Hz) for the β-punicalagin anomer (Fig. S1).
Method validation and quantification of analytes
The UPLC-IMS-HRMS method employed was partially validated in accordance with ICH guidelines [24] by using multiple analytical parameters, namely, method specificity, linearity, precision, and quantification parameters, viz. LOD and LOQ.
Specificity
Analytical standards of punicalin and punicalagin together with the DCM:MeOH extract of G. perpensa and respective blanks was analysed to determine the specificity of the method. Spectral peaks and retention times were used to confirm the identity of the respective peaks. Peak purity was assessed by extracting and comparing the relevant spectra at peak start, peak apex, and peak end. A strong positive correlation was obtained between the DCM:MeOH extract of G. perpensa and the respective ellagitannin standards (Fig. S1-S3).
Linearity, LOD, and LOQ
Linearity of the method was validated by analysing 5 concentrations of each analyte, punicalin and punicalagin. The concentration range used was 1–50 μg/mL for punicalin and 5–80 μg/mL for punicalagin, prepared by serial dilution from a standard stock solution. Triplicate analysis for each analyte was carried out. Strong positive correlation coefficients of 0.9998 and 0.9997 were obtained for the punicalin and punicalagin calibration curves respectively (Table 4; Fig. S4/S5). The calibration curves obtained were of a linear relationship for the tested ranges. The LOD and LOQ for punicalin were found to be 0.27 μg/mL and 0.81 μg/mL. Similarly, the LOD and LOQ for punicalagin were 1.4 μg/mL and 4.1 μg/mL respectively (Table 4), indicating that the method exhibited good sensitivity.
Precision
The precision of the method was evaluated by intra-day tests. The standard solutions at 5 different concentrations were analysed in 3 replicates during the day. To examine the repeatability, 3 different working solutions of the DCM:MeOH extract were analysed (Table 5). Precision and repeatability were expressed as relative standard deviation (%RSD). Values within 5% RSD were deemed acceptable. Intra-day precisions (%RSD) were determined by the measurement of peak area of the reference standard.
Percentage of punicalin and punicalagin in the G. perpensa DCM:MeOH extract and dry plant material
The concentration of punicalin and punicalagin in the DCM:MeOH extract was calculated as 2.12 ± 0.15% and 1.51 ± 0.15% (%w/w), respectively. Additionally, the concentration of punicalin and punicalagin in the dry plant material was calculated as 0.19 ± 0.01% and 0.14 ± 0.01% (%w/w), respectively. The quantity in the dry plant material was determined based on a DCM:MeOH extract, with a 9.2% extraction yield.
Punicalin and punicalagin disrupt RBD/ACE2 interactions in vitro
To demonstrate that punicalin and punicalagin are active constituents in G. perpensa, standards of the 2 compounds were purchased and rescreened for effects on RBD-ACE2 interactions in full-dose response AlphaScreen assays. Both compounds disrupted interactions of the spike glycoprotein RBD with the ACE2 receptor with measured IC50 values of 9 nM and 29 nM observed respectively for punicalin and punicalagin (Fig. 9).

Dose–response curves denoting ability of pure compounds punicalin and punicalagin to disrupt luminescence due to SARS-CoV-2 spike RBD—host ACE2 protein binding in AlphaScreen assay. REGN10987 from Fig. 2 was used as the positive control and graphed for comparison
Discussion
As SARS-CoV-2 cases and COVID-19-related deaths continue to rise globally, development of new therapies and treatments remains crucial. While many antiviral drugs have been developed directly from or based on natural product chemical scaffolds [13], discovery and development of new natural product-based antiviral leads remain limited by the ability to isolate these pure compounds from complex plant extract chemical mixtures. Shifting away from computational and in silico-based screening methods, here we relied on advanced hyphenated analytical techniques to probe G. perpensa L., a medicinal plant reported to be traditionally used for COVID-19 treatment and management in the Eastern Cape, South Africa. The extract, together with its subfractions, was tested for their ability to disrupt the interaction of SARS-CoV-2 spike RBD with the host ACE2 receptor, a key step in SARS-CoV-2 entry and infection. We note that this hypothesis-driven study was based on the traditional claims of its use for treatment of COVID-19, thereby highlighting the importance of reverse pharmacology-based methods which may lead to identification and validation of additional antivirals against SARS-CoV-2 [28]. These leads are particularly relevant as, following further validation and safety assessments, they may provide more accessible options to those in resource-constrained regions where existing therapeutics and vaccines are not readily available or locally accepted.
UPLC-IMS-HRMS was successfully used to confirm the presence of punicalagin and punicalin, 2 ellagitannins, in the DCM:MeOH extract of G. perpensa, with punicalin being previously unreported to occur within the plant. The UPLC-IMS-HRMS method employed was partially validated according to ICH guidelines and the quantity of the two compounds in the DCM:MeOH extract determined with the use of standard calibration curves. The two compounds, punicalin and punicalagin, were found to be present in low concentrations in the bioactive DCM:MeOH extract, specifically 2.12 ± 0.15% and 1.51 ± 0.15% (%w/w) respectively. Based on a DCM:MeOH extraction method with a 9.2% yield, the quantity of punicalin and punicalagin in dry G. perpensa plant material was found to be 0.19 ± 0.01% and 0.14 ± 0.01% (%w/w) respectively. The two compounds, nonetheless, were found to possess potent antagonism against RBD-ACE2 interactions with IC50s in the low nM concentration range. In support of these observations, punicalagin was previously reported to possess antiviral activity against SARS-CoV-2 in plaque reduction assays (EC50 = 7.20 ± 1.08 μM), although the mode of action was proposed to be via inhibition of the viral 3CL protease (MPro) [15]. In contrast, our data clearly show that both punicalagin and punicalin additionally possess RBD-ACE2 antagonistic activity; further mechanistic studies are needed to reconcile these disparate results. As common for natural products, it is possible that punicalin and punicalagin may act on multiple viral proteins to antagonise SARS-CoV-2, which in turn may enhance their antiviral activity and/or set higher genetic barriers to viral resistance. Studies using replication-competent viruses are warranted to elucidate these mechanisms and their contributions to antiviral activity in greater detail.
Ion mobility coupled mass spectrometry is a powerful technique used for the analysis and separation of complex mixtures [29]. Many CCS libraries are becoming available which provide theoretical and/or experimental ion mobility data [30, 31]. These libraries, however, rely on studies such as the one described here to provide reliable experimental data from certified standards. Although no significant differences in CCS nor drift times were observed for the same qualifier quasi-molecular ions of α-punicalagin and β-punicalagin, expectedly, differences in drift time and CCS were observed for the [M-H]− and [M-2H]2− quasi-molecular ions of peaks with the same retention time, i.e. peaks belonging to α-punicalagin or β-punicalagin. With recent advances in IM-MS, like cyclic ion mobility (cIM) [32], differences between anomers would be more significant [33] and would provide added assurance in sample analysis, where anomers might be difficult or impossible to separate with column chromatography. Nonetheless, this finding reinforces the idea that IMS can be used for complex mixtures where a 3rd dimension of separation would be helpful. The study highlights the advantages in combining powerful analytical methods to identify new antiviral chemical leads from complex plant mixtures.
Conclusion
Natural products remain a rich and yet underdeveloped source of drug discovery. As many reported studies rely exclusively on in silico and computational screening of natural products against SARS-CoV-2, this study instead followed an in vitro protein interaction-based approach where potential antiviral properties of G. perpensa were observed. The fractions and pure compounds were identified and isolated through bioassay-guided fractionation, and their exceptional RBD-ACE2 disrupting potential was demonstrated though in vitro bioassays. Additionally, punicalin and punicalagin were quantified in the DCM:MeOH extract. Ion mobility mass spectrometry provides an interesting 3rd dimension of the separation for complex mixtures. Due to the very low concentrations of the ellagitannins in the plant, the hyphenated analytical technologies used provided a means to overcome this difficulty. The results provide new opportunities to probe and investigate medicinal plants and their pure minor chemical components for their activities against SARS-CoV-2. G. perpensa and the 2 ellagitannins identified may also serve as new candidates for eventual SARS-CoV-2 treatments or complementary medicines.
References
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–9. https://doi.org/10.1038/s41586-020-2008-3.
Ibn-Mohammed T, Mustapha KB, Godsell J, Adamu Z, Babatunde KA, Akintade DD, Acquaye A, Fujii H, Ndiaye MM, Yamoah FA, Koh SCL. A critical review of the impacts of COVID-19 on the global economy and ecosystems and opportunities for circular economy strategies. Resour Conserv Recycl. 2021;164. https://doi.org/10.1016/j.resconrec.2020.105169.
Saladino V, Algeri D, Auriemma V. The psychological and social impact of Covid-19: new perspectives of well-being. Front Psychol. 2020;11. https://doi.org/10.3389/fpsyg.2020.577684.
Caniels TG, Bontjer I, van der Straten K, Poniman M, Burger JA, Appelman B, Lavell HAA, Oomen M, Godeke GJ, Valle C, Mögling R, van Willigen HDG, Wynberg E, Schinkel M, van Vught LA, Guerra D, Snitselaar JL, Chaturbhuj DN, Martin IC, Moore JP, de Jong MD, Reusken C, Sikkens JJ, Bomers MK, de Bree GJ, van Gils MJ, Eggink D, Sanders RW. Emerging SARS-CoV-2 variants of concern evade humoral immune responses from infection and vaccination. Sci Adv. 2021;7. https://doi.org/10.1126/sciadv.abj5365.
Kumar S, Maurya VK, Prasad AK, Bhatt MLB, Saxena SK. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). Virus Dis. 2020;31:13–21. https://doi.org/10.1007/s13337-020-00571-5.
Elsawah HK, Elsokary MA, Abdallah MS, ElShafie AH. Efficacy and safety of remdesivir in hospitalized Covid-19 patients: systematic review and meta-analysis including network meta-analysis. Rev Med Virol. 2021;31. https://doi.org/10.1002/rmv.2187.
Kabinger F, Stiller C, Schmitzová J, Dienemann C, Kokic G, Hillen HS, Höbartner C, Cramer P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat Struct Mol Biol. 2021;28:740–6. https://doi.org/10.1038/s41594-021-00651-0.
Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, Boras B, Cardin RD, Carlo A, Coffman KJ, Dantonio A, Di L, Eng H, Ferre R, Gajiwala KS, Gibson SA, Greasley SE, Hurst BL, Kadar EP, Kalgutkar AS, Lee JC, Lee J, Liu W, Mason SW, Noell S, Novak JJ, Obach RS, Ogilvie K, Patel NC, Pettersson M, Rai DK, Reese MR, Sammons MF, Sathish JG, Singh RSP, Steppan CM, Stewart AE, Tuttle JB, Updyke L, Verhoest PR, Wei L, Yang Q, Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;4784:4.
Xiu S, Dick A, Ju H, Mirzaie S, Abdi F, Cocklin S, Zhan P, Liu X. Inhibitors of SARS-CoV-2 entry: current and future opportunities. J Med Chem. 2020;63:12256–74. https://doi.org/10.1021/acs.jmedchem.0c00502.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–3. https://doi.org/10.1126/science.abb2507.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-280.e8. https://doi.org/10.1016/j.cell.2020.02.052.
Mm C. Plant products as antimicrobial agents. Clin Microbiol Rev. 1999;12:564–82.
Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod. 2020;83:770–803. https://doi.org/10.1021/acs.jnatprod.9b01285.
Atanasov AG, Zotchev SB, Dirsch VM, Orhan IE, Banach M, Rollinger JM, Barreca D, Weckwerth W, Supuran CT, et al. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20:200–16. https://doi.org/10.1038/s41573-020-00114-z.
Du R, Cooper L, Chen Z, Lee H, Rong L, Cui Q. Discovery of chebulagic acid and punicalagin as novel allosteric inhibitors of SARS-CoV-2 3CLpro. Antiviral Res. 2021;190: 105075. https://doi.org/10.1016/j.antiviral.2021.105075.
Li H, Xu F, Liu C, Cai A, Dain JA, Li D, Seeram NP, Cho BP, Ma H. Inhibitory effects and surface plasmon resonance-based binding affinities of dietary hydrolyzable tannins and their gut microbial metabolites on SARS-CoV-2 main protease. J Agric Food Chem. 2021;69:12197–208. https://doi.org/10.1021/acs.jafc.1c03521.
Nair MS, Huang Y, Fidock DA, Towler MJ, Weathers PJ. Artemisia annua L. hot-water extracts show potent activity in vitro against Covid-19 variants including delta. J Ethnopharmacol. 2021;284:114797. https://doi.org/10.1016/j.jep.2021.114797.
Tietjen I, Cassel J, Register ET, Zhou XY, Messick TE, Keeney F, Lu LD, Beattie KD, Rali T, Tebas P, Ertl HCJ, Salvino JM, Davis RA, Montaner LJ. The natural stilbenoid (–)-hopeaphenol inhibits cellular entry of SARS-CoV-2 USA-WA1/2020, B.1.1.7, and B.1.351 Variants. Antimicrob Agents Chemother. 2021;65. https://doi.org/10.1128/aac.00772-21.
Mammo FK, Mohanlall V, Shode FO. Gunnera perpensa L a multi-use ethnomedicinal plant species in South Africa. Afr J Sci Technol Innov Dev. 2017;9:77–83. https://doi.org/10.1080/20421338.2016.1269458.
Hutchings A, Scott AH, Lewis G, Bunningham AB. Zulu medicinal plants: an inventory. Pietenmaritzburg: University of Natal Press; 1996.
Thornburg CC, Britt JR, Evans JR, Akee RK, Whitt JA, Trinh SK, Harris MJ, Thompson JR, Ewing TL, Shipley SM, Grothaus PG, Newman DJ, Schneider JP, Grkovic T, O’Keefe BR. NCI program for natural product discovery: a publicly-accessible library of natural product fractions for high-throughput screening. ACS Chem Biol. 2018;13:2484–97. https://doi.org/10.1021/acschembio.8b00389.
Fischer UA, Carle R, Kammerer DR. Identification and quantification of phenolic compounds from pomegranate (Punica granatum L.) peel, mesocarp, aril and differently produced juices by HPLC-DAD-ESI/MSn. Food Chem. 2011;127:807–21. https://doi.org/10.1016/j.foodchem.2010.12.156.
Puljula E, Walton G, Woodward MJ, Karonen M. Antimicrobial activities of ellagitannins against Clostridiales perfringens, Escherichia coli, Lactobacillus plantarum and Staphylococcus aureus. Molecules. 2020;25. https://doi.org/10.3390/molecules25163714.
ICH Validation of analytical procedures text and methodology Q2 (R1) International Conference on Harmonization 2005, Geneva Switzerland. https://doi.org/10.1002/9781118532331.ch23.
Starr TN, Greaney AJ, Addetia A, Hannon WW, Choudhary MC, Dingens AS, Li JZ, Bloom JD. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science. 2021;371:850–4. https://doi.org/10.1126/science.abf9302.
Teixeira N, Nabais P, de Freitas V, Lopes JA, Melo MJ. In-depth phenolic characterization of iron gall inks by deconstructing representative Iberian recipes. Sci Rep. 2021;11:1–11. https://doi.org/10.1038/s41598-021-87969-3.
Lu J, Ding K, Yuan Q. Determination of punicalagin isomers in pomegranate husk. Chromatographia. 2008;68:303–6. https://doi.org/10.1365/s10337-008-0699-y.
Raut AA, Chorghade MS, Vaidya ADB. Reverse pharmacology. Elsevier Inc.; 2017. https://doi.org/10.1016/B978-0-12-801814-9.00004-0.
Lanucara F, Holman SW, Gray CJ, Eyers CE. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem. 2014;6:281–94. https://doi.org/10.1038/nchem.1889.
Zhou Z, Luo M, Chen X, Yin Y, Xiong X, Wang R, Zhu ZJ. Ion mobility collision cross-section atlas for known and unknown metabolite annotation in untargeted metabolomics. Nat Commun. 2020;11:1–13. https://doi.org/10.1038/s41467-020-18171-8.
Goshawk J, Barknowitz G, Mccullagh M. The development of a natural products library using ion - mobility enabled mass spectrometry 2019:2019. https://doi.org/10.13140/RG.2.2.16592.25600.
Giles K, Ujma J, Wildgoose J, Pringle S, Richardson K, Langridge D, Green M. A cyclic ion mobility-mass spectrometry system. Anal Chem. 2019;91:8564–73. https://doi.org/10.1021/acs.analchem.9b01838.
Ujma J, Ropartz D, Giles K, Richardson K, Langridge D, Wildgoose J, Green M, Pringle S. Cyclic ion mobility mass spectrometry distinguishes anomers and open-ring forms of pentasaccharides. J Am Soc Mass Spectrom. 2019;30:1028–37. https://doi.org/10.1007/s13361-019-02168-9.
Acknowledgements
The authors would like to thank Dr. Madelien Wooding for running the samples on the UPLC-IMS-HRMS instrument and for her valuable inputs. Wiehan Rudolph is similarly acknowledged for his valuable inputs. We would also like to acknowledge Magda Nel (Herbarium Assistant at Schweickerdt Herbarium, PRU) for her assistance in plant identification, as well as Dr. Pablo Tebas for the gift of REGN10987 control antibody.
Funding
This research work was supported by the DSI (Department of Science and Innovation Agency of South Africa) and the NRF (National Research Foundation) for student funding. It was also provided by the Wistar Science Discovery Fund (L.J.M. and J.M.S.), the Commonwealth of Pennsylvania (I.T., L.J.M., and J.M.S.), and the Canadian Institutes for Health Research (CIHR PJT-153057) (I.T.). The work was also supported by the following grants to L.J.M.: the Robert I. Jacobs Fund of the Philadelphia Foundation and the Herbert Kean, M.D., Family Professorship.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Freddie J. Isaacs collected and supplied plant material used in study. Luke Invernizzi did data acquisitions, curation, formal analysis, methodology, validation, and writing of the original draft under supervision from Vinesh J. Maharaj and co-supervision from Phanankosi Moyo with assistance from Ian Tietjen. Ian Tietjen: funding acquisition, experiments, formal analysis, validation, and writing of original draft. Joel Cassel: experiments and formal analysis. Joseph M. Salvino: funding acquisition and writing of manuscript. Luis J. Montaner: funding acquisition and writing of manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Invernizzi, L., Moyo, P., Cassel, J. et al. Use of hyphenated analytical techniques to identify the bioactive constituents of Gunnera perpensa L., a South African medicinal plant, which potently inhibit SARS-CoV-2 spike glycoprotein–host ACE2 binding. Anal Bioanal Chem 414, 3971–3985 (2022). https://doi.org/10.1007/s00216-022-04041-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-022-04041-3