
Abstract
Remdesivir is a nucleotide analog prodrug that has received much attention since the outbreak of the COVID-19 pandemic in December 2019. GS-441524 (Nuc) is the active metabolite of remdesivir and plays a pivotal role in the clinical treatment of COVID-19. Here, a robust HPLC-MS/MS method was developed to determine Nuc concentrations in rat plasma samples after a one-step protein precipitation process. Chromatographic separation was accomplished on Waters XBrige C18 column (50 × 2.1 mm, 3.5 μm) under gradient elution conditions. Multiple reaction monitoring transitions in electrospray positive ion mode were m/z 292.2 → 163.2 for Nuc and 237.1 → 194.1 for the internal standard (carbamazepine). The quantitative analysis method was fully validated in line with the United States Food and Drug Administration guidelines. The linearity, accuracy and precision, matrix effect, recovery, and stability results met the requirements of the guidelines. Uncertainty of measurement and incurred sample reanalysis were analyzed to further ensure the robustness and reproducibility of the method. This optimized method was successfully applied in a rat pharmacokinetics study of remdesivir (intravenously administration, 5 mg kg−1). The method can act as a basis for further pharmacokinetic and clinical efficacy investigations in patients with COVID-19.

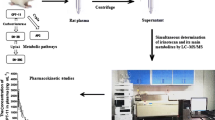
Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the beginning of the global COVID-19 health crisis in December 2019, 124.5 million people have been infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). As of Jun 22, 2021, 3,872,457 people have died from COVID-19. Owing to this global emergency, remdesivir has attracted attention because of its potential as a clinical COVID-19 therapy. Remdesivir is a nucleotide analog prodrug that is intracellularly metabolized to an analog of adenosine triphosphate. It has a broad-spectrum effect against several viruses, such as Ebola, SARS-CoV, MERS-CoV, and SARS-CoV-2, in vitro and in vivo [1]. Coronaviruses can cause many serious diseases, such as acute respiratory distress syndrome, multisystem organ failure, and, ultimately, death [2]. 1′-Cyano-substituted adenine C-nucleoside ribose analog (GS-441524, Nuc) is the active metabolite of remdesivir and plays a critical role in inhibiting viral RNA synthesis [3]. Multiple clinical trials have been conducted to investigate the therapeutic effectiveness of remdesivir for COVID-19 [4, 5].
However, to the best of our knowledge, no analytical high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method or pharmacokinetic application is available for the quantitation of Nuc in rat plasma. A comprehensive understanding of the pharmacokinetics and pharmacodynamics of remdesivir is indispensable for improving its risk-benefit ratio in COVID-19 patients. Although Warren et al. [6] reported a pharmacokinetic profile of remdesivir in healthy rhesus macaques, the detailed LC-MS/MS method and its validation results are inadequate for preclinical and clinical applications. The HPLC-MS/MS method published by Avataneo et al [7] for the determination of remdesivir and GS-441524 (Nuc) required a large volume of plasma (50 μL) and had low extraction recovery (67%). This method could not satisfy the clinical quantitation to some extent, where patients are so weak that they cannot tolerate large blood draws. Thus, there is no appropriate detailed method available with the potential for clinical application.
Owing to the severity of the current global pandemic and the emerging need for a robust clinical method to assess blood concentrations/exposure, a selective, reliable, and reproducible HPLC-MS/MS assay was firstly developed and fully validated for quantitating Nuc, the active metabolite of remdesivir, in plasma. Furthermore, this method was successfully applied in a pharmacokinetics study in rat. The findings reported here may act as a reference for future antiviral drug development and pharmacokinetic investigations in preclinical and clinical studies.
Experimental
Materials and reagents
Remdesivir and Nuc (>98% pure) were obtained from DC Chemicals Company (Shanghai, China). Carbamazepine (internal standard, IS, lot: 100142-201706, 100% pure) was obtained from the National Institutes for Food and Drug Control (Beijing, China). Ammonium formate, methanol (MeOH), acetonitrile (ACN), and isopropanol (IPA) (all HPLC grade) were purchased from Fisher Scientific (Boston, MA) and were used to prepare solutions and mobile phases. Solution modifier of HPLC-grade formic acid was obtained from DIKMA Co., Inc. (China). The rat control sodium fluoride/potassium oxalate (NaF/K-Ox) plasma was obtained from healthy rats. A Milli-Q purification system was used to produce ultrapure water.
HPLC-MS/MS for quantitation
A liquid chromatography system (LC-20ADXR, Shimadzu, Japan) was adopted for the chromatographic separation. The autosampler was set up at room temperature, and the column temperature was maintained at 40 °C. The flow rate was fixed at 0.4 mL min−1 and the injection volume was 1 μL. Chromatographic separation was accomplished on a Waters XBrige C18 column (50 × 2.1 mm i.d., 3.5 μm; Waters, Milford, USA) with a run time of 4.5 min. Mobile phase A (ACN:water = 95:5, v/v, 0.1% formic acid) and phase B (water:ACN = 99:1, v/v, 0.1% formic acid) were freshly prepared. The following gradient elution program was used to obtain the baseline separation: 1.2 min, 99% B; 1.2–3.5 min, 5% B; 3.5–3.6 min, 99% B; 3.6–4.5 min, 99% B. Methanol:water (1:1, v/v) was used to prevent for potential carryover.
A mass spectrometer (QTRAP 5500, SCIEX, Canada) equipped with a positive Turbo Ionspray electrospray ionization source was employed to in the present study [8]. The mass spectrometry parameters were set as follows: ion-spray voltage, 4500 V; gas temperature, 650 °C; curtain gas, 20.0 psi; ion source/turbo gas, 30.0 psi; cell exit potential, 30.0 V. Protonated molecule [M+H]+ ion was used for all analytes. Twin multiple reaction monitoring (MRM) transitions were used for the quantifier and qualifier. The quantitative MRM was set at m/z 292.2 → 163.2 for Nuc, and 237.1 → 194.1 for the IS. Raw data and chromatography data were acquired and analyzed using Analyst coupled with MultiQuant 3.0 (version 1.6.3 and 3.0.3, SCIEX).
Preparation of standard solutions, calibrations samples, and quality control samples
Stock solutions of remdesivir, Nuc, and the IS were made in MeOH to final concentrations of 1.0 mg mL−1. The analytes were weighed twice to prepare the stock solutions of the standards and quality control (QC) samples at concentrations of 500 μg mL−1. A working IS solution (2 ng mL−1 in MeOH) was freshly produced prior to batch analysis. Working calibration solutions of Nuc (1 mg mL−1) were prepared in MeOH:water = 1:1 (v/v) at concentration of 20, 50, 100, 200, 500, 1000, 2000, 5000, and 10,000 ng mL−1. Similarly, working QC solutions with analytes (1 mg mL−1) were prepared at concentration of 20, 50, 100, 1000, and 8000 ng mL−1 in MeOH:water = 1:1 (v/v). The calibration and QC solutions were prepared individually. All solutions were kept at −80 °C before analysis.
Calibration standards were prepared in drug-free rat plasma by diluting the corresponding working solutions 10-fold to achieve nominal concentrations of 2, 5, 10, 20, 50, 100, 200, 500, and 1000 ng mL−1. Similarly, QC plasma samples were obtained using blank rat plasma at nominal analyte concentrations of 2, 5, 10, 100, and 800 ng mL−1.
Preparation of plasma samples for quantitative analysis
All plasma samples were vortexed for about 1 min, and then, a 20-μL aliquot was transferred to a 1.5-mL centrifuge tube. The samples were extracted via protein precipitation. A 100-μL aliquot of IS working solution (2 ng mL−1) was added, and the proteins were quickly precipitated by vortexing for approximately 30 s. The samples were centrifuged at 14, 000 rpm for 10 min at 4 °C, and the supernatant was diluted with ACN:water (1:9, v/v) at a ratio of 1:3. The diluted extraction solutions were transferred to autosampler vials. A 1-μL aliquot was injected for HPLC-MS/MS analysis.
Method validation
Biological method validation of the specificity, linearity, accuracy, precision, matrix effect, recovery, and stability was performed in accordance with the US Food and Drug Administration guidelines [9] and the China Pharmacopeia (version 2015) [10].
Specificity and selectivity were assessed using double blank (e.g., without Nuc or IS), blank (IS), lower limit of quantification (LLOQ; the lowest concentration of the calibration curve with an accuracy and precision ≤ ± 20% of the nominal value and a signal-to-noise ratio ≥ 5), and unknown samples to assess the chromatographic integrity and possible disturbances in the retention time of analytes.
Carryover is a challenge in the quantitative analysis of biological samples in complex matrices. After analysis of the upper limit of quantification (ULOQ) standard, three consecutive aliquots of double blank samples were injected and evaluated by comparing the peak areas. The observed peak areas were ≤ 20% of the LLOQ, indicating the absence of carryover [11].
The concentrations of LLOQ, low QC1 and QC2 (LQC1 and LQC2), medium QC (MQC), and high QC (HQC) for Nuc were 2, 5, 10, 100, and 800 ng mL−1, respectively. Five different QC concentrations (n = 6) were prepared to assess the accuracy and precision in three different batches. The acceptable criteria for accuracy and precision were %bias ≤ ± 15% and coefficient of variance (%CV) <15%.
Two sets of calibration standards were analyzed for each analytical batch—one set at the beginning and one set at the end of the batch [8]. Weighted linear regression (1/x2) and the squared correlation coefficient (r) were calculated, and r ≥ 0.990 was accepted for indicating linearity.
The recovery of analyte and IS expressed as percentage was investigated by comparing peak areas of the LQC, MQC, and HQC (n = 6) sample concentrations in which plasma was added to the analyte before and after the extraction. IS recovery was also performed in accordance with a previous sample extraction procedure [8].
According to our previous evaluation method [8], matrix effects (MEs) were assessed by adding known amounts of standard solutions at three concentrations (LQC, MQC, and HQC) to the extracted samples. Drug-free plasma (n = 6) was employed and spiked with the neat solutions. Two types of peak areas were compared—spiked plasma post-extraction (A) and water-substituted samples (B)—at equivalent concentrations. The ME was calculated as (A/B × 100). Data were considered to be acceptable when the inter-sample variability range was within 15% [11].
The following stability parameters were evaluated at the LQC and HQC concentrations (n = 6) with an acceptability criteria of %bias ≤15%: ambient temperature short-term stability (24 h), three sequential freeze-thaw cycles (−80 °C to ambient temperature), post-preparation stability (left in the autosampler for 24 h), and long-term stability (−80 °C for 3 months).
Uncertainty of measurement
Measurement uncertainty is associated with the robustness and reliability of results, and is an estimated value [12]. This value characterizes the range of the true value of a measurement. The bottom-up approach was used to assess the measurement uncertainty in accordance with a previous study [12]. Both standard uncertainty which comprises type A uncertainty (evaluation by statistical analysis of observations) and type B uncertainty (evaluation by means other than statistical analysis of observations) and expanded uncertainty (k = 2, 95% confidence) were evaluated to reveal measurement uncertainty. When estimating measurement uncertainty, the analysis process must be clearly defined to determine the performance characteristics of the equipment and the source and impact of the uncertainty. Basic sources of uncertainty were considered in this study, such as volumetric operations, weighing, the purity of standards and reference materials, atomic and molecular weights, multiple-point calibration (linear regression), and recovery. The samples at concentration of 100 ng mL−1 (n = 18, three groups) were analyzed to evaluate measurement uncertainty of the HPLC-MS/MS method [13].
Pharmacokinetic study in rats
Sprague-Dawley rats (male, 180–220 g, 8 weeks) were obtained from Beijing Vital River Laboratory Animal Technologies Co. Ltd (Beijing, China) and acclimated for 7 days in an environmentally controlled room (25 ± 2 °C, 55 ± 5% relative humidity, 12-h light cycle). Animal care was conducted in accordance with the Guidelines for Animal Experimentation of Capital Medical University (Beijing, China) and the protocol was approved by the Animal Ethics Committee of the institution.
After fasting for 12 h, six rats were intravenously administered remdesivir (5 mg kg−1) dissolved with 12% sulfobutylether-β-cyclodextrin in water. Blood samples (approximately 500 μL) were collected from the ophthalmic veins into (NaF/K-Ox) tubes before administration and at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, and 48 h after administration. All samples were directly centrifuged at 3500 rpm for 10 min at 4 °C. The separated plasma was frozen at −80 °C.
Incurred sample reanalysis (ISR) was carried out to verify the accuracy and precision established with the QCs samples. Eleven incurred samples near the Cmax and in the elimination phase were reanalyzed in a separate batch. The data were considered acceptable when the concentration obtained from the reanalysis was within 20% of the initial analysis mean for at least 67% of the repeats [9, 13].
WinNonlin (Pharsight 8.3, Mountain View, CA) with non-compartmental model was used to calculate the pharmacokinetic parameters. The analytical parameters including the maximum concentration (Cmax), time to Cmax (Tmax), area under the plasma concentration-time curve (AUC0-t), and half-life (t1/2) were assessed as previously described [8].
Results and discussion
Optimization of the mass spectrometry detection conditions
To optimize the mass spectrometry conditions, needle pump injection was used to obtain the largest analyte responses. Precursor ions of Nuc and the IS exhibited the best MS responses. Based on our observations, the qualitative and quantitative detection performed well by ESI+ and MRM. The product ion fragmentation patterns are shown in Fig. 1. All analytes and the IS gave predominantly single-charged protonated precursors [M+H]+ at m/z 292.2 → 163.2 (quantitative ion) and 292.2 → 147.2 (qualitative ion) for Nuc, and 237.1 → 194.1 for IS (Fig. 1). Additionally, the accuracy of MRM determination was ensured by using a twin ion, and the product ions were sufficiently stable for reproducible quantitation.

MRM transitions of Nuc (A) and IS (B) in ESI+ mode
Optimization of the chromatographic conditions
The analytes and IS were used to optimize the HPLC conditions. Various chromatography columns were initially tested and compared. Nuc showed a broadening peak on the Waters Symmetry C18 column (50 × 2.1 mm, 5 μm) when ACN was used as the organic phase. This broad peak was modified on the Waters XBrige C18 column (50 × 2.1 mm, 3.5 μm) when chromatographic separation was accomplished under gradient elution condition, leading to peak shapes with improved separation, symmetry, and sensitivity. MeOH and ACN were evaluated as better chromatographic solutions. The peak shape and response were significantly improved when using ACN; thus, ACN was selected as the organic phase. The retention time of Nuc was considerably reduced when pure acetonitrile and pure water were applied as the mobile phase. Optimization of the composition and pH resulted in an optimal organic/water phase of ACN/water. To improve the intensity of the analytes and IS, different concentrations of formic acid and acetic acid were tested. After careful comparison, 0.1% formic acid was added to the organic/water phase. The column temperature was assessed at room temperature, 40 °C and 55 °C. After considering the responses and separation efficiency, a column temperature of 40 °C was adopted for the HPLC-MS/MS method. The autosampler was set at room temperature. Generally, a smaller injection volume leads to a smaller residual amount of analyte in the LC-MS/MS system. When optimizing the liquid chromatography conditions, we used an injection volume of 1 μL, and the system contained almost no residue. A small injection volume is also beneficial for multiple analyses in case of instrument failure, environmental emergencies, or limited post-preparation solutions. Related to potential carryover or residue, the analyte response was too high; therefore, we reduced the injection volume to avoid column saturation.
Isocratic and gradient elutions were both examined for the elution mode. Based on the elution results, gradient elution was selected and optimized to enhance the separation. The entire analytical run time was 4.5 min. Several flow rates (0.1–0.7 mL min−1) were tested to optimize the performance, and a flow rate of 0.4 mL min−1 was found to be the optimum speed. Analogs or stable isotope-labelled internal standards are appropriate for the determination of analytes because they have the same LC conditions as the analyte of interest. In the current study, carbamazepine as the IS was suitable because it satisfied the quantitative analysis as shown in term of specificity, matrix effect, and recovery. Baseline separation was obtained, and the retention time was 1.31 min for Nuc, and 1.77 min for the IS (Fig. 2).

Typical chromatography of Nuc and IS in rat plasma
In the process of optimizing the LC conditions, carryover was found when quantitating the low-concentration samples. Thus, three washing solution were investigated: MeOH:water = 1:1 (v/v), ACN:water = 1:1 (v/v), and MeOH:ACN:IPA:water = 1:1:1:1 (v/v/v/v). Carryover was successfully eliminated when using MeOH:water = 1:1 (v/v) as a washing solution.
Sample preparation
Three extraction methods were assessed during method development and validation: protein precipitation (PPT), liquid-liquid extraction (LLE), and solid-phase extraction (SPE). PPT is a simple, high-throughput, and energy-efficient method, especially for complex biological matrixes. Warren et al. [6] previously reported a three-step PPT method using 25 μL of plasma sample. After precipitation, evaporation, and reconstitution, a 10-μL aliquot was analyzed by LC-MS/MS; however, the extraction recovery of this method was not described in detail in the study. Avataneo et al. [7] developed an HPLC-MS/MS method using 50 μL of plasma; however, this larger sample volume is not suitable for patients with serious COVID-19. In the present study, a simple one-step PPT method using 20 μL of plasma was adopted for sample preparation. To achieve linearity of the standard curve while retaining an applicable method, the supernatants were further diluted after precipitation, and a volume of 1 μL was injected for analysis. The recovery and accuracy/precision results also indicated that the PPT procedure satisfied the quantitative determination requirements for Nuc (Table 1). Compared with the published assays, this simple, high-recovery, one-step protein precipitation method was better able to satisfy the demands for determining the tested analytes.
Method validation for quantitation
Figure 2 shows a representative 4.5-min chromatography run. Selectivity and specificity are important considerations of an HPLC-MS/MS method. Samples from six different rats were used to investigate the selectivity by assessing double blank, blank, LLOQ, and unknown plasma samples. As shown in Fig. 3, there was no interference peak or crosstalk at the retention time when using the optimized HPLC-MS/MS conditions, indicating the selectivity and specificity of the method.

Chromatograms of Nuc (A) and IS (B) in double blank plasma without analyte and IS, blank plasma with IS, blank plasma with Nuc at the LLOQ with IS, and unknown sample after administration of 5 mg kg−1 dose of remdesivir (41.3 ng mL−1 for Nuc; 4 h after administration)
The accuracy and precision (Table 1) results suggested that the LLOQ of 2 ng mL−1 for Nuc met the requirements of this quantitative test. Moreover, the Nuc limit of detection was 1 ng mL−1 at a signal-to-noise of S/N ≥ 5.
Carryover was evaluated to ensure that it did not affect the precision or accuracy of the HPLC-MS/MS method. During the validation procedure, carryover was observed for the double blank plasma sample following the ULOQ. Several preventive procedures were implemented to eliminate the effect of carryover on quantitation, such as arranging the sample sequence order from low to high concentration, inserting double blank samples after the high-concentration samples, and reducing the injection volume. Regrettably, none of these actions reduced the carryover. Next, various solutions (MeOH:water (1:1, v/v), ACN:water (1:1, v/v), and MeOH:ACN:IPA:water (1:1:1:1, v/v/v/v)) for washing the injection needle were compared with respect to the carryover peak, accuracy, and integrity. The MeOH:water (1:1, v/v) needle wash solution was found to eliminate carryover, and an acceptable level of accuracy and quantitative determination was achieved.
The accuracy and precision were evaluated based on three different batches of five QC concentrations. The accuracy and precision values were all within ±4.98% and 9.55% for the intra- and inter-day analyses, respectively (Table 1). All data met the requirement for biological sample analysis, indicating that the method was robust and accurate.
The linearity of Nuc was assessed using the IS. After optimization, the Nuc calibration curve achieved a range 2–1000 ng mL−1 with a coefficient correlation (r) of greater than 0.990. The representative calibration equation for Nuc was y = 0.00403x + 0.00886 (r = 0.99587), where y represents the peak area ratio of analyte to IS and x represents the concentration of analytes in rat plasma.
As shown in Table 2, after the simple one-step precipitation, the mean recovery of Nuc was constant within the range of 77.47–95.27% with a %CV of ≤12.84%. For the matrix effect (indicated by the matrix factor), the range of IS-normalized matrix factors was 94.30–109.60% with a CV% of less than 13.09%. These results demonstrated that the analysis was robust.
The stability results of the low- and high-concentration QC samples are presented in Table 3. The accuracy and precision were all within the ±15% criteria. Nuc in rat plasma was stable under several storage conditions: at room temperature for 4 h, during post preparation in the autosampler for 24 h, during three consecutive freeze-thaw cycles (−80 °C to room temperature), and during long-term storage at −80 °C for 3 months. Previous studies have shown that the prodrug remdesivir has relatively poor stability in mouse plasma (half-life <5 min) owing to the presence of a secreted carboxylesterase 1c (Ces1c), which is absent in humans [14, 15]. Thus, measures are needed to reduce remdesivir degradation during method validation, rat blood extraction and plasma generation, and shipping. Adding a plasma esterase enzyme inhibitor to the plasma or placing the samples under “heat stress” may help to improve the instability of remdesivir. Overall, the stability results obtained in the current study were consistent with those from previous studies [7, 14, 15].
Uncertainty of measurement
When quantitatively assessing a biological matrix, understanding the uncertainty of measurement can ensure that the results are fit for the intended purpose while also helping to explain the results [16]. As shown in Fig. 4, the measurement uncertainty rose from uncertainties in the repeatability, sample weighing, solution preparation, sample extraction and recovery, HPLC-MS/MS system, and calibration curve. At a concentration of 100 ng mL−1, the largest amount of uncertainty was from the calibration curve, accounting for 65% of the total uncertainty. Uncertainty arising from the HPLC-MS/MS error accounted for the smallest amount (0.02%). The final amount of measurement uncertainty at a Nuc concentration of 100 ng mL−1 was 101.82 ± 23.97 ng mL−1 (mean ± expanded uncertainty), with a coverage factor of k = 2 (95% confidence). These results indicate that when preparing the standard curve, particular attention should be focused on the state of the plasma, establishing the linear range and pre-treatment of the biological sample to minimize the measurement uncertainty.

Uncertainty of measurement (A) and proportion (B) for determining Nuc at concentration of 100 ng mL−1
Pharmacokinetics and incurred sample reanalysis
This method was successfully applied in a pharmacokinetic study in rats after intravenous administration of remdesivir (Fig. 5). Remdesivir is a monophosphoramidate prodrug of an adenosine analog (Nuc). Remdesivir has played a pivotal role in the treatment of MERS-CoV, SARS-CoV, and SARS-CoV-2 [3, 14, 17, 18]. After intravenous administration, remdesivir is rapidly converted in the blood into the active metabolite Nuc. Figure 5 shows the mean plasma concentration-time profile. Upon administration, remdesivir exhibited a short plasma half-life (t1/2 = 0.39 h), and the active metabolite (Nuc) efficiently reached the sites where virus may persistent. The pharmacokinetic parameters of Nuc are shown in Table 4. Owing to the rapid conversion to Nuc, the median Tmax was 0.5 h, and the t1/2 was 5.48 ± 2.54 h. To further elucidate the pharmacokinetic parameters, several pharmacokinetics studies in rat and other species were comprehensively assessed [6, 19,20,21]. However, only two studies investigated the pharmacokinetic parameters of Nuc rather than remdesivir [20, 21]. Furthermore, different doses were administered, and the drug formulation also varied. The pharmacokinetic parameters of Nuc shown in Table 4 were comparable with previous findings [20]. The results of the present study, such as the t1/2, Cmax, and AUC values, will provide a basis for further clinical dosage or blood sampling investigations.

The mean concentration-time curve of Nuc in rat after intravenous administration of remdesivir at dose of 5 mg kg−1 (n = 6) as well as graphical representation of incurred sample reanalysis results
In the present study, the LLOQ of remdesivir was set at 1 ng mL−1 and had acceptable accuracy and precision. This optimized HPLC-MS/MS method was sensitive enough to determine the plasma concentration up to 48 h after administration. The mean pharmacokinetic profile and pharmacokinetic parameters of Nuc are shown in Fig. 5 and Table 4, respectively.
The robustness and reliability of the HPLC-MS/MS method were tested using 11 incurred samples. Except for the two samples, all other changes were ≤ ±20% of the initial analysis, which meets the bioanalytical method validation guideline criteria [9].
Although the reliable and simple HPLC-MS/MS method developed in this study was applied successfully to investigate rat pharmacokinetics, the ultimate objective of this quantitation assay is to determine the concentrations of remdesivir and Nuc in humans. Limitations of this study are the instability of remdesivir, the lack of thorough measures to avoid or reduce remdesivir degradation, and the absence of more challenging stability studies (e.g., Ces1c inhibitor, heat stress). Future investigation should include such parameters, which could aid in resolving the COVID-19 pandemic situation. In our future work, we will continue to optimize the developed assay to overcome these limitations, with the aim of addressing the therapeutic urgency of COVID-19.
Comparing the present method to other reported assays
Recently, Alvarez et al. [22] developed an HPLC-MS/MS method and applied it to assess a patient who was intravenously administered remdesivir. However, the required plasma volume was 50 μL and matrix effects were not reported. Moreover, detailed recovery, uncertainty of measurement, and incurred sample reanalysis were not determined. A comparison of the method reveals several advantages of the present HPLC-MS/MS method, such as simple plasma preparation, satisfactory matrix effect and recovery, detailed pharmacokinetic application, and robustness owing to measuring the uncertainty and incurred sample reanalysis (Table 5).
Conclusions
A high-throughput, robust, sensitive, and reproducible HPLC-MS/MS method was developed and fully validated for the quantitation of Nuc, the active metabolite of remdesivir, in plasma. Worsening of the COVID-19 pandemic has necessitated the development of an HPLC-MS/MS method that can accurately quantitate Nuc to provide plasma exposure information, because obtaining this information has become a bottleneck in the clinical treatment of COVID-19. The optimized HPLC-MS/MS method reported here offers significant advantages including cost-effective, high-throughput sample preparation (protein precipitation extraction), and requiring only small volume of biological matrix (20 μL). Furthermore, this method meets the requirements for extraction recovery and matrix effects. This quantitative method was also successfully applied in a rat pharmacokinetics study. This reliable method will provide sufficient reference and mirror for pharmacokinetics and clinical efficacy in patients with COVID-19.
References
Lamb YN. Remdesivir: first approval. Drugs. 2020;80(13):1355–63.https://doi.org/10.1007/s40265-020-01378-w.
Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, et al. Compassionate use of remdesivir for patients with severe Covid-19. New Engl J Med. 2020;382(24):2327–36. https://doi.org/10.1056/NEJMoa2007016.
Brown AJ, Won JJ, Graham RL, Dinnon KH 3rd, Sims AC, Feng JY, et al. Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antivir Res. 2019;169:104541. https://doi.org/10.1016/j.antiviral.2019.104541.
Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569–78. https://doi.org/10.1016/S0140-6736(20)31022-9.
Amirian ES, Levy JK. Current knowledge about the antivirals remdesivir (GS-5734) and GS-441524 as therapeutic options for coronaviruses. One Health. 2020;9:100128. https://doi.org/10.1016/j.onehlt.2020.100128.
Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531(7594):381–5. https://doi.org/10.1038/nature17180.
Avataneo V, de Nicolo A, Cusato J, Antonucci M, Manca A, Palermiti A, et al. Development and validation of a UHPLC-MS/MS method for quantification of the prodrug remdesivir and its metabolite GS-441524: a tool for clinical pharmacokinetics of SARS-CoV-2/COVID-19 and Ebola virus disease. J Antimicrob Chemother 2020;75(7):1772–7. https://doi.org/10.1093/jac/dkaa152.
Du P, Guan Y, An Z, Li P, Liu L. A selective and robust UPLC-MS/MS method for the simultaneous quantitative determination of anlotinib, ceritinib and ibrutinib in rat plasma and its application to a pharmacokinetic study. Analyst. 2019;144(18):5462–71. https://doi.org/10.1039/c9an00861f.
Evaluation, Research CFD. Center for Drug Evaluation and Research of the U.S. Department of Health andHuman Services Food and Drug Administration. Guidance for industry: bioanalytical method validation. https://www.fda.gov/downloads/Drugs GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf 2018 [updated 2018-01-01%\ 2020-05-11 11:19:00.
State PC. People’s Republic of China Pharmacopoeia. Part IV 2015 [updated 2015-01-01. 363–8].
Du P, Han X, Li N, Wang H, Yang S, Song Y, et al. Development and validation of an ultrafiltration-UPLC-MS/MS method for rapid quantification of unbound docetaxel in human plasma. J Chromatogr B. 2014;967(15):28–35. https://doi.org/10.1016/j.jchromb.2014.07.007.
Meyer VR. Measurement uncertainty. J Chromatogr A. 2007;1158(1–2):15–24. https://doi.org/10.1016/j.chroma.2007.02.082.
Du P, Li P, Zhao R, Liu H, Liu L. Optimized UPLC-MS/MS method for the quantitation of olanzapine in human plasma: application to a bioequivalence study. Bioanalysis. 2019;11(13):1291–302. https://doi.org/10.4155/bio-2019-0114.
Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017;9(396):eaal3653. https://doi.org/10.1126/scitranslmed.aal3653.
Li B, Sedlacek M, Manoharan I, Boopathy R, Duysen EG, Masson P, et al. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem Pharmacol. 2005;70(11):1673–84. https://doi.org/10.1016/j.bcp.2005.09.002.
JCGM 100, Evaluation of measurement data – guide to the expression of uncertainty in measurement with minor corrections (GUM), www.bipm.org, (2008).
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–71. https://doi.org/10.1038/s41422-020-0282-0.
de Wit E, Feldmann F, Cronin J, Jordan R, Okumura A, Thomas T, et al. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci U S A. 2020;117(12):6771–6. https://doi.org/10.1073/pnas.1922083117.
Hu W-j, Chang L, Yang Y, Wang X, Xie Y-c, Shen J-s, et al. Pharmacokinetics and tissue distribution of remdesivir and its metabolites nucleotide monophosphate, nucleotide triphosphate, and nucleoside in mice. Acta Pharmacol Sin. 2020:1–6.
Li Y, Cao L, Li G, Cong F, Li Y, Sun J, et al. Remdesivir metabolite GS-441524 effectively inhibits SARS-CoV-2 infection in mouse models. J Med Chem. 2021.
National Center for Advancing Translational Sciences. https://opendata.ncats.nih.gov/covid19/GS-441524. (accessed 2020-12-20).
Alvarez J-C, Moine P, Etting I, Annane D, Larabi IA. Quantification of plasma remdesivir and its metabolite GS-441524 using liquid chromatography coupled to tandem mass spectrometry. Application to a Covid-19 treated patient. Clin Chem Lab Med. 2020;58(9):1461–8. https://doi.org/10.1515/cclm-2020-0612.
Acknowledgements
We thank Guoyong Wang for supplying standards of remdesivir and Nuc. We thank Katherine Thieltges from Liwen Bianji, Edanz Editing China (Edanz) (www.liwenbianji.cn/acwww.liwenbianji.cn/), for editing the English text of a draft of this manuscript.
Funding
This research was in part supported by the National Natural Science Foundation of China (81703611), Beijing Municipal Administration of Hospitals’ Youth Program (QML20180305), Beijing Municipal Natural Science Foundation (7164262), and Scientific Research Foundation of Capital Medical University (PYZ20022).
Author information
Authors and Affiliations
Contributions
Ping Du: conceptualization, methodology, HPLC-MS/MS method, writing (original draft). Guoyong Wang: investigation, editing. Song Yang: investigation, writing (review and editing). Pengfei Li: quality control, pharmacokinetic experiment, writing (review and editing). Lihong Liu: conceptualization, validation, quality control, writing (review and editing), supervision.
Corresponding authors
Ethics declarations
Ethics approval
All animal studies in this project including their welfare adhered to the guidelines of the Animal Experimentation of Capital Medical University (Beijing, China), and complied to the “National Research Council’s Guide for the Care and Use of Laboratory Animals.” The protocol was approved by the Animal Ethics Committee of the Institution.
Consent to participate
Not applicable
Consent for publication
All authors listed have approved the enclosed manuscript.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Du, P., Wang, G., Yang, S. et al. Quantitative HPLC-MS/MS determination of Nuc, the active metabolite of remdesivir, and its pharmacokinetics in rat. Anal Bioanal Chem 413, 5811–5820 (2021). https://doi.org/10.1007/s00216-021-03561-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-021-03561-8