
Abstract
A novel, generally applicable method of identifying peptides using HPLC, microwave-assisted acid hydrolysis (MAAH), and bioinformatics is described. Method validation was performed on bacteriocins—antibacterial peptides produced by probiotic bacteria—using nine different bacteriocin isolates secreted by the probiotic Lactococcus lactis. Calibration curves were constructed for 23 amino acid PTH derivatives, and analysis was performed using norleucine as the internal standard. Validation of amino acid analysis performed in the range 2.5–100 nmol/mL indicated excellent method linearity, while the LODs ranged from 0.17 to 2.88 nmol/mL and the LOQs from 0.51 to 8.75 nmol/mL. The MAAH method was developed by irradiating nisaplin for various durations at 700 W, with 7 min providing the best results. The amino acid content of each sample was estimated following the application of MAAH to ten different samples. The bacteriocins in our samples were identified using the UniProt database. Eight of nine peptides were identified as UniProt entries: nisin A (P13068), nisin Z (P29559), I4DSZ9, OB7236, P36499, OB7237, A0A0M7BH60, and T2C9F0. The phylogenetic tree was constructed for nisin A and nisin Z using the multiple sequence aligning tool Clustal Ω. The identified nisin types presented excellent correlation with their ModBase-predicted structures. The present method gives true, precise, and rapid results, and requires only standard technical equipment. Our results suggest that the present approach can facilitate the discovery of novel bacteriocins and provide useful information on not only the amino acid contents of peptides but also the evolution of protein biology.

Identification of eight bacteriocins secreted by the probiotic L. lactis, following microwave assisted acid hydrolysis (MAAH), amino acid content analysis of each sample with HPLC-DAD and bioinformatics analysis using Uniprot, Clustal Ω and ModBase.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Antibiotic resistance resulting from the development of multidrug-resistant (MDR) pathogens is a major problem in animal and human medicine, and has prompted an urgent drive to discover and characterize novel antibacterials [1]. One interesting class of natural antibacterials comprises the compounds produced by probiotic bacteria, which are characterized as bacteria beneficial to the host [2, 3]. Given the prominent role of the gut microbiome in the survival of an organism [4], the advantage of using probiotics as a preventive measure is well documented [5, 6]. Bacterial balance in the gastrointestinal tract is achieved through quorum sensing, which includes the production of bacteriocins [7]. Bacteriocins are primary metabolites produced by lactic acid bacteria (LAB), which are generally recognized as safe (GRAS) [8]. These peptides act as food preservatives [9], even at picomolar levels [10], as well as antibacterials against animal and human pathogens [11, 12]. Bacteriocins are synthesized ribosomally and they form a heterogeneous group of compounds that present considerable diversity in terms of their structure and properties [1]. Various bacteriocin classification schemes have been suggested, comprising either two [12], three [13], or four [14] groups. However, class I bacteriocins (known as lantibiotics) and class II bacteriocins (nonlantibiotics) are common to all of the classifications; the main difference between these classes is that lantibiotics contain post-translationally modified residues such as β-lanthionine and 3-methyllanthionine, which help to stabilize the peptides with respect to heat, pH, and proteolysis [15].
The bacteriocin that has received FDA and EU approval for use in food destined for human consumption is nisin [16]. Nisin is produced by Lactococcus lactis and is main representative of the family of lantibiotics. Nisin A, has been described as a 3500-Da elongated amphipathic peptide that acts by producing pores on the pathogen’s membrane [17]. Moreover, L. lactis bacteriocins have been successfully applied against animal pathogens [18, 19]. Recently, a screening method for the simultaneous quantification of nine bacteriocins produced by L. lactis was reported [20]. However, these bacteriocins were not identified as their amino acid contents were not determined, and it is well accepted that the amino acid composition is an important peptide classification parameter [21]. Amino acid analysis is an indispensable protein analysis technique that consists of two steps, namely the preparation of the protein hydrolysate and the subsequent quantitative analysis of the amino acids that are formed [22]. The rate-limiting and most important step in this procedure has been identified as the preparation of protein hydrolysates [23]. To this end, as an alternative to the commonly used yet time-consuming method of constant-boiling hydrochloric acid [24], the faster and more efficient technique of microwave-assisted acid hydrolysis (MAAH) has been introduced [25]. This method has been used extensively for the identification of transmembrane proteins [26] and for the mapping of protein sequences and their modifications [27], while its use in combination with microwave-assisted base hydrolysis (MABH) to generate ladder sequence information has also been reported [28].
The analysis of the resulting amino acids has been performed following their derivatization with phenylisothiocyanate (PITC) as phenylthiohydantoin (PTH) derivatives [29], dansyl derivatives (either alone or in combination with the Edman procedure) [30], or o-phthalaldehyde derivatives [27]. However, analytical approaches employing reverse-phase instead of ion exchange chromatography of the PITC-derivatized amino acids [31, 32] have allowed the efficient separation and quantification of amino acids in various proteins.
The selection of the candidate probiotic, the isolation of proteins from the cell-free medium, their hydrolysis, followed by amino acid content determination is the classical or empirical method employed for bioactive peptide discovery. However, bioinformatics-driven approaches that exploit information in various databases to assign particular biological activities to isolated peptides have recently been introduced [33, 34]. Indeed, the screening of anticancer bacteriocins in the human gut microflora has been performed using such an approach [35]. In fact, in-silico approaches such as genome mining and peptide sequencing are now considered indispensable for the analysis of antibiotic biosynthetic pathways [36].
Although the nisins produced by L. lactis have gained extensive attention, other antibacterial peptides produced by this probiotic bacterium may also prove to be of great value. Since the discovery of novel bacteriocins is important for enhancing the arsenal of available antimicrobials, an efficient screening system for them appears to be required. In the work discussed in the present paper, a system combining the classical method of analysis using MAAH with bioinformatics for the screening and identification of the nine bacteriocins produced by L. lactis ATCC 11454 is presented.
Materials and methods
Chemicals and reagents
All amino acids and Nisaplin® (N 5764) were purchased from Sigma–Aldrich (Taufkirchen, Germany). The internal standard norleucine, the derivatization reagent PITC, and DL-dithiothreitol were purchased from Alfa Aesar (Karlsruhe, Germany). HPLC-grade methanol, acetonitrile, trifluoroacetic acid (TFA), sodium acetate, sodium dihydrogen phosphate, and disodium hydrogen phosphate were purchased from Merck (Darmstadt, Germany). Triethylamine (TEA) was purchased from Fisher (Loughborough, UK) and absolute ethanol was from Panreac (Montcada i Reixac, Spain).
A stock solution of each amino acid was prepared by dissolving 0.5 mmol of each amino acid into 50 mL of coupling buffer, resulting in a final amino acid concentration of 10 mM, except for norleucine, which amounted to 50 mM. The coupling buffer consisted of ethanol (EtOH), triethylamine (TEA), and H2O in the ratio 1:1:1. Stock solutions of all the standard amino acids were prepared, as well as those of some modified forms, making a total of 24 amino acid solutions. Stock solutions were stored at 4 °C for a period of up to 1 month.
Sample preparation and derivatization
All of the standard amino acids, as well as some modified forms, were derivatized according to the Edman derivatization process [29]; the protocol published by Checa-Moreno et al. was used [32], albeit with some modifications. Briefly, 250 μL of each amino acid and an equal amount of the internal standard norleucine were dissolved in the coupling buffer and submitted to solvent evaporation in a rotary evaporator (Buchi, Flawil, Switzerland). The dry residue was allowed to react with 200 μL of the derivatization solution, which consisted of EtOH, H2O, TEA, and phenyl isothiocyanate (PITC) at a ratio of 6:2:2:1, for 10 min at room temperature under a hood. It then underwent solvent evaporation to dryness in a rotary evaporator, and the amino acid derivatives were collected in 1 mL of buffer consisting of (1.25 mM Na2HPO4–5 mM NaH2PO4, pH 7.4)–6% (v/v) ACN. The 1-mL derivative solution with a final concentration of 2.5 mM was stored at 4 °C in 1.5-mL Eppendorf reaction tubes, or at −20 °C for long-term storage, which did not have any significant negative effects.
HPLC analysis
The HPLC apparatus consisted of a Shimadzu (Kyoto, Japan) LC20AD pump equipped with a quaternary solvent delivery system, a Vydac C18 (4.6 × 250 mm, 5 μm; Hichrom, Reading, UK) reverse-phase column, a column oven, and an SPD-20A diode array detector (Shimadzu). The HPLC-DAD was controlled by a computer employing the LC solution data acquisition software supplied by Shimadzu. In the gradient elution, mobile-phase component A was (sodium acetate 0.28 M, pH 6.7)–5% acetonitrile (ACN)–0.05% triethylamine (TEA), while mobile-phase component B was ACN–H2O 60:40 (v/v), and the oven was set to 40 °C. The sample volume of the valve injector was 80 μL. The gradient started with 100% A and 0% B, after which B was increased stepwise to 40% at 24 min, 50% at 25 min, 80% at 26 min, and 90% at 30 min; this was held until the end of analysis. The analysis time was 35 min. After changing back to 100% solvent A at the end of analysis, the mobile phase was maintained for 10 min to achieve column equilibration before the next injection. The flow rate was 0.5 mL/min during the whole gradient cycle, and the column temperature was set to 40 °C. The analytes were monitored at 220, 280, and 254 nm, and absorption spectra (190–800 nm) of the analytes were recorded throughout all analyses.
Method validation
Calibration curves of the amino acid derivatives were constructed by injecting samples into the HPLC system to obtain six calibration points for each derivative. The concentrations used for this purpose were 2.5, 5, 25, 50, 75, and 100 nmol mL−1, while the final concentration of the derivatized internal standard norleucine was 12.5 nmol mL−1. For each calibration curve, linear regression statistical analysis was performed, employing the regression data analysis package of Microsoft Excel. In each case, we determined the standard curve equation (y = ax + b) along with the correlation coefficient R 2, the limit of detection (LOD), the limit of quantification (LOQ), the recovery rate, and the accuracy in terms of trueness (bias %) and precision (intraday and interday). LOD was calculated as 3.3 × (SEintercept/slope), and the quantification limit LOQ as 10 × (SEintercept/slope). Trueness was calculated as bias (%) = (c − n)/n × 100%, where c is the calculated value for the standard and n is its nominal value. Intraday precision was calculated using the RSD% of at least three replicates at each calibration level daily, while interday precision was calculated using the RSD% of six replicates over two days. The extraction recoveries of the 24 derivatives were determined by comparing the concentration of each amino acid estimated from the calibration curve (c) to its nominal concentration (n) as (c/n) × 100%.
Microwave-assisted acid hydrolysis (MAAH)
In this study, we attempted the microwave-assisted acid hydrolysis of 10 potential bacteriocin samples isolated in previous experiments in our lab [20]. Each sample was lyophilized to discard the buffer used in the previous experiment and resuspended in 1 mL of phosphate buffer (1.25 mM Na2HPO4–5 mM NaH2PO4) pH 7.4 containing 6% (v/v) ACN. A volume of 10 μL of each sample was transferred to an empty 1.5-mL reaction tube to which 10 mL of 20 mM of dithiothreitol (DTT) solution was added to prevent the oxidization of the amino acids [26]. After 20 min of incubation of the aforementioned mix at 60 °C, 20 or 40 μL of 50% v/v trifluoroacetic acid (TFA) were added. Then the contents of the reaction tube with the new mix were exposed to nitrogen gas to remove atmospheric oxygen. The sample was centrifuged at 3800×g for 30 s in a bench centrifuge. Then the capped lid was covered with Teflon tape and the reaction tube was pinned in a polystyrene holder floating on 200 mL ddH2O in a 600-mL beaker. The beaker was exposed to microwaves for 5, 7, 10, or 15 min at 700 W. Following centrifugation (30 s, 3800×g), the hydrolyzed sample underwent solvent evaporation to dryness in a rotary evaporator and then derivatization according to the method described above. Nisin was used as a MAAH optimization control when performing MAAH protocol validation. Briefly, a stock solution (1 mg/mL) of nisin was purified from Nisaplin® [37], which contains 2.5% nisin (106 IU/g). A volume of 10 μL of this stock solution was used throughout the MAAH procedure described above. The hydrolyzed nisin was derivatized and injected in order to select the best protocol by comparing the amino acid profile of the nisin with a corresponding profile obtained from the literature. The protocol employing 40 μL TFA 50% (v/v), a microwave irradiation time of 7 min, and the use of nitrogen gas was identified as the most efficient.
Computational analysis of bacteriocins of L. lactis
Bioinformatics tools and databases were employed to achieve a better understanding of the antimicrobial peptide secretome of L. lactis. The sequences of L. lactis bacteriocins were obtained via the UniProt database [38]. Multiple sequence alignment was performed and the phylogenetic tree of the sequences was acquired using the multiple sequence aligning tool Clustal Ω 1.2.1 [39]. The 3D structures of nisin A and Z were obtained from the Protein Data Bank (PDB) [40], while possible structure models were taken from ModBase [41].
Results and discussion
Standard curves of amino acid derivatives
The retention times and LOQ and LOD values for the 23 amino acids examined are shown in Table 1. All amino acid analyses provided LOD values of <1 nmol/mL except for glycine, alanine, cystine, and lysine derivatives. This may be attributed to the wide range of concentrations (2.5–100 nmol/mL) employed to construct these standard curves, a fact that led to high error values at the lowest concentration. In the standard curve for the cysteine derivative, more than one peak was present in the chromatogram (Table 1); in that case, the first peak (at 16.3 min, Table 1), which had the biggest peak area, was considered in the calculations. The production of cysteine derivatives when using the PITC derivatization method has been reported, so cysteine is often not included in the determination of PTH derivatives of amino acids [32]. However, the fact that our approach based on the largest peak was not realistic enough led us to include the cysteine derivative when attempting to estimate the amino acid compositions of the hydrolyzed bacteriocins.
Although slight differences were observed between the retention times for the multiderivative mixes and those for the single-derivative runs, the elution sequence remained the same (Table 1). However, the relative difference (%) of the relative retention time of each derivative compared to the corresponding value for the MAAH optimization control was lower than 2.5% with the exception of tyrosine and valine (with values of 2.6 and 2.8%, respectively). In the sample chromatogram of a mix containing individually derivatized amino acids (Fig. 1a), each derivative was identified by considering the retention time as well as the peak shape, absorption strength, and the coelution pattern for multiple amino acids. In our preliminary experiments, the application of a steeper gradient of 80% B at 24 min resulted in the coelution of leucine, isoleucine and norleucine. This issue was circumvented by introducing two more steps into our gradient (namely 40% B at 24 min, 50% at 25 min, and 80% at 26 min), which were subsequently used throughout our experiments.

Representative chromatogram of a the mix containing all of the independently derivatized amino acids (excluding cysteine) and b nisin hydrolyzed using the MAAH method, after exposure to microwaves for 7 min. In a, there is twice as much glycine (Gly) as the concentration calculated for identification purposes. Cst cystine
The standard curve equations are presented alongside the correlation factors (R 2) and validation data in Table S1 of the “Electronic supplementary material” (ESM). Our data demonstrate that there are excellent correlations in most cases, with the exception of the derivatives of leucine, isoleucine, lysine, and glycine (R 2 < 0.9990). Recovery values ranged from 90 to 115%, with the exception of the lowest recoveries observed at the low calibration point (2.5 nmol/mL) for isoleucine (86.66%) and leucine (87.06%) and the peak value of 116.0% for glycine. Moreover, the corresponding standard curves presented lower R 2 values, possibly due to a narrower range of linearity. The bias (%) and the intraday and interday precision (RSD %) were lower than 10% with the exception of the low calibration points for glycine, alanine, and leucine (Table S1 in the ESM). The calculations used to determine the method bias, precision, and recovery are described in the “Method validation” section.
MAAH protocol optimization
To optimize the MAAH protocol, we employed the well-studied peptide nisin as a MAAH optimization control, enabling us to establish the optimal hydrolysis conditions. This peptide has previously been used in derivatization experiments employing dansyl chloride to determine the relative content of polypeptide chains [30], but (to the best of our knowledge) not in any MAAH protocols. In the present study, a variety of microwave exposure times were examined. Initially, 10 and 15 mins of exposure time were tested. When the absorption signals of the samples hydrolyzed for 10 min are compared to those of the samples hydrolyzed for 15 min, the latter signals appear to be attenuated compared to the former (see Fig. S1b, c in the ESM). In particular, the peak at 13.128 min for the sample exposed to 10 min of irradiation appears to be augmented compared to the peak at 12.584 min in the sample irradiated for 15 min. In addition, a few obvious peaks in the chromatogram of the sample irradiated for 10 min (e.g., the peak at 11.170 min) do not correspond to any of those in the chromatogram for the sample irradiated for 15 min. In general, there are only a few peaks that are likely to correspond to amino acid derivatives, which is not enough to allow the confident identification of a specific peptide from among a bunch of bacteriocins.
An exposure time of 5 min was then tested to determine if it would be adequate for peptide hydrolysis. This led to incomplete hydrolysis (Fig. S1a in the ESM), as many peaks with high absorption values eluted at 25–28 min, which most probably correspond to peptide fractions of the initial bacteriocin. It has been reported [21] that 50% TFA (20 μL) and 20 min of irradiation were used for the mapping of proteins including casein and albumin. Likewise, for membrane proteins such as bacteriorhodopsin, a combination of 25% v/v TFA and 10 min of irradiation provided the best results [26]. Therefore, 7 min was tested as an intermediate exposure time between 5 min and 10 min, while the volume of 50% v/v TFA was doubled to a final volume of 40 μL (Fig. 1b). These conditions were considered optimal in terms of the number of peaks that correspond to potential amino acid derivatives in the hydrolyzed sample. In this manner, and given the number of amino acid residues in nisin, the number of peaks that probably correspond to amino acid derivatives was deemed adequate to recognize the peptide.
Another point of interest was whether exposure of the hydrolysis mix to nitrogen gas would improve the output from hydrolysis by removing the oxidizing agent, namely oxygen. Treating the samples with nitrogen led to more peaks in the chromatogram of the derivatized hydrolysate (Fig. S2 of the ESM), possibly due to the oxidation of amino acid derivatives in the absence of nitrogen. An example is provided by methionine, which exhibits a peak with a retention time of 21.451 ± 0.11 for the hydrolysate (Table 1) that is absent when the nitrogen step is omitted (Fig. S2 in the ESM). Hence, the nitrogen gas exposure step was included for all of the hydrolysis mixes to suppress oxidation of the individual amino acid residues during the hydrolysis process.
Amino-acid profiling of L. lactis bacteriocins
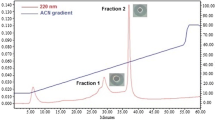
The bacteriocins of L. lactis that were profiled in the present study originated from an experimental setup employed previously by our group [20]. Briefly, the bacteriocins were initially precipitated with ammonium sulfate from the cell-free growth medium of L. lactis, leading to sediment 1, while further precipitation of the supernatant with organic solvents (ethanol:diethyl ether, 1:2 v/v) produced sediment 2. Both sediments presented antimicrobial activity, and in the final step of reverse-phase preparative chromatography, a strong signal was observed with the same retention time as and an identical absorption spectrum to pure nisin. The bacteriocin samples (1–10) that were profiled are displayed in Fig. 2, where each bacteriocin corresponds to a peak in the relative chromatogram (sample 1 was a blind blank that did not present antibacterial activity and was used for validation purposes). Our approach takes into account the total concentration of each recognizable amino acid that is included in each sample of the same peptide, and the major criterion employed is the ratio of the amino acid concentrations, which is compared with the corresponding concentration ratio of already known amino acids. As the dataset used was not extensive, emphasis was placed on the amino acid residues that showed high interpeptide variation among the bacteriocins secreted by the bacterium. The known amino acid compositions of all the bacteriocins obtained from UniProt are provided in the ESM (Tables S2–S11). The amino acid compositions of each of our samples were compared with the data in those tables for identification purposes. As expected, there were small deviations in our calculations that may be due to the variety of strains of L. lactis that were included in the UniProt results. The sample recognition process as well as possible explanations for certain deviations that were observed are addressed in the ESM. Each of the ten samples was annotated with a UniProt entry ID, as shown in Fig. 2, apart from samples 1 and 10, for which peptide recognition was not feasible. This was expected for the blind blank (sample 1), but sample 10 has been reported to present antibacterial activity [20] and thus requires further investigation, as it may represent a bacteriocin that has not been registered yet.

Samples 1–10 were isolated in previous experiments and correspond to specific retention times in each chromatogram: 1: 20–21 min, 2: 21–22 min, 3: 22–23 min, 4: 24–25 min, 5: 26–27 min, 6: 27–28 min, 7: 29–30 min, 8: 29–30 min, 9: 30–31 min, 10: 31–32 min. Each sample is annotated with a UniProt entry ID, apart from samples 1 and 10, for which peptide recognition was not feasible
We only discuss samples 7 and 8 in detail below, under the assumption that they correspond to nisins A and Z, respectively. In sample 7 (sediment 1, 29–30 min), as shown in Fig. 3a, the following were detected: [Asp] =0.94 nmol/mL, [Asn] = 1.67 nmol/mL, [Gly] = 36.23 nmol/mL, [His] = 24.32 nmol/mL, [Met] = 4.71 nmol/mL, [Val] = 5.39 nmol/mL, [Ile] = 121.59 nmol/mL. This sample is proposed to represent nisin A, based on previously obtained retention time and absorbance spectrum data [20]. The present work offers additional support for this proposal: the ratio [Asp]/[Asn], the high concentrations of Thr and Ile, and the similar [Met]/[Val] ratio. Larger peaks at 12.3 min and 24.55 min probably correspond to small peptides that were not hydrolyzed. However, since such unhydrolyzed peptides were not present in the pure nisin hydrolysate (Fig. 1b), these were attributed to possible contamination from the previous peak during sample collection in preparative HPLC [20]. The peak preceding sample 7 did not present antimicrobial activity, meaning that it does not relate to a bacteriocin and may not have been hydrolyzed under the MAAH conditions used in the present study.

Chromatograms obtained following MAAH hydrolysis. a Sample 7 (Asp: 5.781 min, Asn: 7.843 min, Gly: 9.079 min, His: 10.552, Thr: 15.145 min, Met: 21.504 min, Val: 21.860 min, Ile: 25.961 min, NOR: 29.272 min). b Sample 8 (Asn: 7.982, Gly: 9.077 min, His: 11.118 min, Met + Val: 22.196 min, Ile: 26.124 min, Leu: 27.424 min, NOR: 29.505 min)
In sample 8 (sediment 2, 29–30 min), shown in Fig. 3b, the amino acid concentrations were estimated as [Asn] = 2.58 nmol/mL, [Gly] = 35.45 nmol/mL, [His] = 14.97 nmol/mL, [Ile] = 104.99 nmol/mL, [Leu] = 135.93 nmol/mL. The ratio [Ile]/[Leu] is pretty similar to the corresponding one from Table S5 of the ESM. The larger peaks at 12.8, 18.5, and 24.8 min were attributed to contaminating unhydrolyzed peptides originating from the previous HPLC fraction, as also seen for sample 7 (Fig. 3a). In addition, the increased Asn concentration and the lower His concentration compared to sample 7 provide further evidence that sample 8 might be nisin Z. However, single amino acid differences are hard to validate based on chromatography alone. For this reason, we performed a further validation experiment in which ESI-MS was applied to samples 7 and 8 (Fig. S11 in the ESM). Thus, by examining peaks in the m/z range 500–1200, it was possible to estimate molecular weights of 3354 for sample 7 and 3330.4 for sample 8, which are in good agreement with the reported values for nisins A and Z, respectively [42].
Computational validation of the L. lactis bacteriocin isolation experiments
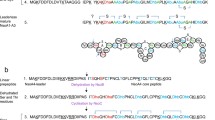
Although nisin is present in both of the samples described above, the question of whether these are different forms of nisin requires investigation. The L. lactis strain ATCC 11454 used in the present study has been reported to produce the main variants of nisin, namely nisin A and nisin Z [43]. In fact, six natural forms of nisin produced by Lactococcus lactis species have been identified: nisin A, nisin Z, nisin Q, and nisin F, U and U2 [44]. After searching the protein database UniProt for the proteins secreted by L. lactis, 12 peptide sequences were retrieved. Employing the multiple sequence alignment tool (Clustal Ω), we were able to identify certain conserved motifs (Fig. 4), and amino acid sequence predictions were used to construct the corresponding phylogenetic tree showing the coevolution of these peptides (Fig. 5). Nisin A is probably the evolutionary progenitor of nisin Z. The sequences of these two peptides have exactly the same number of amino acid residues and they differ in only one single amino acid at position 50, where nisin A has a histidine (H) residue while nisin Z has an asparagine residue (N). Following the original identification of nisin Z as a natural nisin variant [45], the commercially available Nisaplin® was characterized as nisin A [46]. This slight difference between the two peptides leads to significant structural differences: according to the UniProt data, nisin Z has a helix and turn that are absent from nisin A (Fig. 5). Moreover, the ModBase predictions for the structures of these peptides (Fig. 6) suggest that nisin Z has multiple folds that are not present in nisin A. It should be noted that the prediction statistics do not lead to reliable structure models, but they can highlight general trends for the structures. The folding differences between the two forms of nisin suggest that nisin A is slightly more hydrophobic. This rough conclusion may in fact lend further support to our assumption that the nisin-related peaks in the chromatograms obtained after the two precipitation methods (Fig. 2), as previously reported [20], correspond to different types of nisin. The fact that aqueous solvents with high ionic strengths are preferred for the precipitation of hydrophilic molecules while less polar solvents are used to precipitate hydrophobic molecules suggests that nisin A is present in sample 7 (originating from sediment 1 following ammonium sulfate precipitation) and nisin Z is present in sample 8 (originating from sediment 2 following precipitation with organic solvents). This assumption was further validated using the MS data for the two samples (Fig. S11 in the ESM). The slightly acidic pH of the ammonium sulfate solution (pH ~ 5.5) due to the hydrolysis of the salt, in combination with the fact that nisin Z has been reported to be more soluble than nisin A at pH values above 6 (due to the higher polarity of the Asn residue than the His residue in nisin A [46]), provides further support for our hypothesis. Likewise, nisin A and nisin Z were recently reported to yield different extraction patterns, which were attributed to their composition [47].

Multiple sequence alignment with Clustal Ω. The conserved motifs are those in the boxes

Phylogenetic tree of the peptides of interest. Nisins A and Z are annotated. The sequence alignment of these two peptides is given, while structural features (a helix and turn) are highlighted in the nisin Z sequence

Models for the 3D structures of nisins A and Z from ModBase
The secretion of multiple nisins appears to be an evolutionary adaptation of L. lactis to multiple environments. The production of multiple bacteriocins from one bacterium is considered a strategy that allows the bacterium to control competing bacteria through nutrient and space antagonism [48]. Hence, in order to conquer different habitats, L. lactis has enriched its antimicrobial secretome machinery at the minor cost of a single amino acid change to a bacteriocin.
Conclusions and future perspectives on the application of this method
An important dataset was developed in this work: the standard curves of the PTH derivatives of all possible amino acids. These allow us to precisely estimate the amino acid compositions of certain peptides (containing up to 100 residues) that have been hydrolyzed. The MAAH method is highly recommended, as it is far faster than traditional hydrolysis methods and it is precise and well suited to such studies. For proteins or protein complexes (sequences of >100 residues), an initial semi-complete hydrolysis and then complete hydrolysis of the fractions until they are identified may be advisable. The computational tools employed in our study undoubtedly played a prominent role not only in the final identification of the bacteriocins of L. lactis but also when validating our previous experiments and assumptions. It should be noted that it was possible to use these simple computational tools in our work because we had a nonextensive dataset of target peptides to identify. The procedure presented in this paper, based on MAAH hydrolysis and computation tools, is an inexpensive and fast peptide recognition method, and we recommend its use either alone or in combination with other methods of analysis.
References
Perez RH, Zendo T, Sonomoto K. Novel bacteriocins from lactic acid bacteria (LAB): various structures and applications. Microb Cell Factories. 2014;13(Suppl. 1):S3.
Fuller R. Probiotics in human medicine. Gut. 1991;32(4):439–42.
Hill C, Guarner F, Reid G, Gibson G, Merenstein D, Pot B, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11(8):506–14.
Round JL, Mazmanian SK. The gut microbiome shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–23.
Mombelli B, Gismondo MR. The use of probiotics in medical practice. Int J Antimicr Agents. 2000;16:531–6.
Waikar Y. Review of probiotics in children. Pediatr Infect Dis. 2013;5:9–12.
Ohland CL, Jobin C. Microbial activities and intestinal homeostasis: a delicate balance between health and disease. Cell Mol Gastroenterol Hepatol. 2015;1(1):28–40.
USFDA. GRAS notice no. GRN 000064; 2000. Washington, DC: U.S. Food and Drug Administration.
Chikindas ML, Weeks R, Drider D, Chistyakov VA, Dicks LMT. Functions and emerging applications of bacteriocins. Curr Opin Biotech. 2017;49:23–8.
Woraprayote W, Malila Y, Sorapukdeeb S, Swetwiwathana A, Benjakul S, Visessanguan W. Bacteriocins from lactic acid bacteria and their applications in meat and meat products. Meat Sci. 2016;120:118–32.
Touraki M, Karamanlidou G, Karavida P, Karamanoli C. Evaluation of the probiotics Bacillus subtilis and Lactobacillus plantarum bioencapsulated in Artemia nauplii against vibriosis in European sea bass larvae (Dicentrarchus labrax, L.). World J Microbiol Biotechnol. 2012;28(6):2425–33.
Cotter PD, Ross RP, Hill C. Bacteriocins—a viable alternative to antibiotics? Nature Rev Microbiol. 2013;11:95–105.
Ahmad V, Khan MS, Jamal QMS, Alzohairy MA, Al Karaawi MA, Siddiqui MU. Antimicrobial potential of bacteriocins: in therapy, agriculture and food preservation. Int J Antimicrobial Agents. 2017;49:1–11.
McAuliffe O, Ross RP, Hill C. Lantibiotics: structure, biosynthesis and mode of action. FEMS Microbiol Rev. 2001;25(3):285–308.
Nishie M, Nagao JI, Sonomoto K. Antibacterial peptides “bacteriocins”: an overview of their diverse characteristics and applications. Biocontrol Sci. 2012;17(1):1–16.
WHO. Specification for the identity and purity of food additives and their toxicological evolution. 12th Report of the Joint FAO/WHO Expert Committee on Food Additives. WHO technical report series no. 430. Geneva: World Health Organization; 1969.
Klaenhammer TR. Bacteriocins of lactic acid bacteria. Biochimie. 1988;70:337–49.
Vaseeharan B, Ramasamy P. Control of pathogenic Vibrio spp. by Bacillus subtilis BT23, a possible probiotic treatment for black tiger shrimp Penaeus monodon. Lett Appl Microbiol. 2003;36:83–7.
Touraki M, Karamanlidou G, Koziotis M, Christidis I. Antibacterial effect of Lactococcus lactis subsp. lactis on Artemia franciscana nauplii and Dicentrarchus labrax larvae against the fish pathogen Vibrio anguillarum. Aquacult Int. 2013;21:481–95.
Katharopoulos E, Touloupi K, Touraki M. Monitoring of multiple bacteriocins through a developed dual extraction protocol and comparison of HPLC-DAD with turbidometry as their quantification system. J Microbiol Methods. 2016;127:123–31.
Wang N, Li L. Reproducible microwave assisted acid hydrolysis of proteins using a household microwave oven and its combination with LC-ESI MS/MS for mapping protein sequences and modifications. J Am Soc Mass Spectrom. 2010;21:1573–87.
Fountoulakis M, Lahm HW. Hydrolysis and amino acid composition analysis of proteins. J Chromatogr A. 1998;826:109–34.
Chiou SH, Wang KT. Peptide and protein hydrolysis by microwave irradiation. J Chromatogr B. 1989;491:424–31.
Hirs CHW, Stein WH, Moore S. The amino acid composition of ribonuclease. J Biol Chem. 1954;211:941–50.
Chen SH, Chiou SH, Chu YH, Wang KT. Rapid hydrolysis of proteins and peptides by means of microwave technology and its application to amino acid analysis. Int J Peptide Protein Res. 1987;30:572–6.
Zhong H, Marcus SL, Li L. Microwave-assisted acid hydrolysis of proteins combined with liquid chromatography MALDI MS/MS for protein identification. J Am Soc Mass Sprectom. 2005;16:471–81.
Wang N, Li L. Reproducible microwave-assisted hydrolysis of proteins using a household microwave oven and its combination with LC-ESI MS/MS for mapping of protein sequences and modifications. J Am Soc Mass Sprectom. 2010;21:1573–87.
Reiz B, Li L. Microwave-assisted acid and base hydrolysis of intact proteins containing disulfide bonds for protein sequence analysis by mass spectrometry. J Am Soc Mass Sprectom. 2010;21:1596–605.
Edman P, Högfeldt E, Sillén L, Kinell P. Method for determination of the amino acid sequence in peptides. Acta Chem Scand. 1950;4:283–93.
Spivak VA, Levjant MI, Katrukha SP, Varshavsky JAM. Quantitative Ultramicroanalysis of amino acids in the form of their DNS derivatives. Anal Biochem. 1971;44:503–18.
Henrickson RI, Meredith SC. Amino acid analysis by reverse phase high performance liquid chromatography: precolumn derivatization with phenylisothiocyanate. Anal Biochem. 1984;136:65–74.
Checa-Moreno R, Manzano E, Mirón G, Capitán-Vallvey L. Revisitation of the phenylisothiocyanate-derivatives procedure for amino acid determination by HPLC-UV. J Sep Sci. 2008;31(22):3817–28.
Capriotii AL, Cavaliere C, Piovesana S, Samperi R, Lagana A. Recent trends in the analysis of bioactive peptides in milk and dairy products. Anal Bioanal Chem. 2016;408:2677–85.
Gabere MN, Noble WS. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics. 2017;33(13):1921–9.
Duy Nguyen V, Nguyen HHC. Molecular screening of azurin-like anticancer bacteriocins from human gut microflora using bioinformatics. In: Le Thi H, Nguyen N, Do T, eds. Advanced computational methods for knowledge engineering. Advances in Intelligent Systems and Computing, vol 358. Cham: Springer; 2015.
Weber T. In silico tools for the analysis of antibiotic biosynthetic pathways. Int J Med Microbiol. 2014;304:230–5.
Gut M, Prouty AM, Ballard JD, van der Donk WA, Blanke SR. Inhibition of Bacillus anthracis spore outgrowth by nisin. Antimicrob Agents Chemother. 2008;52(12):4281–8.
UniProt Consortium. UniProt. 2016. Available at: http://www.uniprot.org. Accessed 9 Aug 2016.
EMBL-EBI. Clustal Omega. 2016. Available at: https://www.ebi.ac.uk/Tools/msa/clustalo/. Accessed 9 Aug 2016.
Berman H. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–42.
Pieper U, Webb B, Dong G, Schneidman-Duhovny D, Fan H, Kim S, et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2013;42(D1):D336–46.
Zendo T, Nakayama J, Fujita K, Sonomoto K. Bacteriocin detection by liquid chromatography/mass spectrometry for rapid identification. J Appl Microbiol. 2008;104:499–507.
Beasley SS, Saris PEJ. Nisin-producing Lactococcus lactis strains isolated from human milk. Appl Env Microbiol. 2004;70(8):5051–3.
Field D, Connor PMO, Cotter PD, Hill C, Ross RP. The generation of nisin variants with enhanced activity against specific gram-positive pathogens. Mol Microbiol. 2008;69(1):218–30.
Mulders JW, Boerrigter IJ, Rollema HS, Siezen RJ, de Vos WM. Identification and characterization of the lantibiotic nisin Z, a natural nisin variant. Eur J Biochem. 1991;201(3):581–4.
Matsusaki H, Sonomoto K, Ishizak A. Some characteristics of nisin Z, a peptide antibiotic produced by Lactococcus lactis IO-1. Food Sci Technol Int Tokyo. 1998;4(4):290–4.
Ko KY, Park SR, Lim HS, Park SJ, Kim M. Improved pretreatment method for determination of nisins A and Z and monitoring in cheese using liquid chromatography-tandem mass spectrometry. Food Anal Methods. 2016;9:122–30.
Egan K, Field D, Rea MC, Ross RP, Hill C, Cotter PD. Bacteriocins: novel solutions to age old spore-related problems? Front Microbiol. 2016;7:1–21.
Acknowledgements
We thank Sarli Vasiliki, Assistant Professor at the Laboratory of Organic Chemistry, Department of Chemistry, A.U.TH., for the ESI-MS runs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study did not receive any funding other than state funding of the university. The authors declare that they have no conflict of interest.
The present study does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 414 kb)
Rights and permissions
About this article

Cite this article
Tasakis, R.N., Touraki, M. Identification of bacteriocins secreted by the probiotic Lactococcus lactis following microwave-assisted acid hydrolysis (MAAH), amino acid content analysis, and bioinformatics. Anal Bioanal Chem 410, 1299–1310 (2018). https://doi.org/10.1007/s00216-017-0770-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0770-3