
Abstract
Accurate quantification of plasma glucagon levels in humans is necessary for understanding the physiological and pathological importance of glucagon. Although several immunoassays for glucagon are available, they provide inconsistent glucagon values owing to cross-reactivity of the antibodies with peptides other than glucagon. To overcome this limitation, we developed a novel method to measure glucagon levels by a liquid chromatography (LC)-high-resolution mass spectrometry (HRMS) assay via parallel reaction monitoring (PRM) without immunoaffinity enrichment. Using stable isotope-labeled glucagon as an internal standard and 200 μL of plasma, the lower limit of quantification was 0.5 pM. This method was applied to measure plasma glucagon levels during the oral glucose tolerance test (OGTT) and meal tolerance test (MTT) in healthy volunteers, and its results were compared with those of sandwich enzyme-linked immunosorbent assay (ELISA) and radioimmunoassay (RIA). During the OGTT, this method showed significant suppression of plasma glucagon levels, and similar patterns were observed with sandwich ELISA and RIA. In contrast, during the MTT, plasma glucagon levels were slightly elevated according to the LC-MS/MS and sandwich ELISA results and were reduced according to RIA results. Our newly developed LC-MS/MS method overcomes a lack of specificity among currently available immunoassays for glucagon and may contribute to a better understanding of the importance of glucagon.

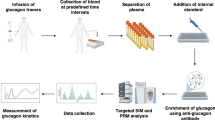
Flowchart for the extraction and quantification of glucagon in human plasma, and plasma glucagon responses in healthy volunteers quantified by the present LC-MS/MS, sandwich ELISA, and RIA during OGTT and MTT
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucagon is a 29-amino acid peptide hormone mainly released from pancreatic alpha cells, although it is also secreted from the stomach and small intestine. It was originally revealed that glucagon (glucagon1–29) is released in response to low blood glucose levels to break down glycogen and stimulate gluconeogenesis in the liver. However, Mighiu et al. [1] showed that hypothalamic glucagon signaling inhibits hepatic glucose production. Thus, glucagon does not simply oppose the effects of insulin. Glucagon is now being investigated as a novel target for the treatment of diabetes, as many studies have suggested that an excess of glucagon, rather than a deficiency of insulin, is the essential characteristic of diabetes [2]. Therefore, accurate measurement of plasma glucagon concentrations in healthy people and diabetes patients is essential to elucidate the physiological and pathophysiological roles of glucagon.
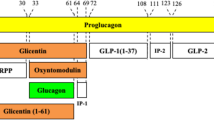
Since the first immunoassay-based method was developed in 1959 [3], immunochemical approaches remain the most common methods for the quantification of plasma glucagon levels, including radioimmunoassays (RIAs) and enzyme-linked immunosorbent assays (ELISAs). Although many immunoassay-based methods have been developed, they suffer from a lack of specificity. Glucagon and other proglucagon-derived peptides are produced by tissue-specific post-translational processing (Fig. 1) [4]. The amino acid sequences of some of these peptides overlap with that of glucagon, resulting in cross-reactive antibodies that cause a lack or reduction of specificity in immunoassay-based methods. Bak et al. [5] compared eight different commonly used glucagon immunoassays and reported that none provided sufficient specificity for glucagon measurement; they concluded that there is a clear need for improved glucagon assays. Importantly, Holst et al. [6] proposed that glucagon can only be measured using the sandwich ELISA. Albrechtsen et al. [7] compared three sandwich ELISA systems (Meso Scale Discovery, Millipore, and Mercodia) with regard to specificity, precision, and sensitivity and concluded that the Mercodia sandwich ELISA showed the best performance. However, Matsuo et al. [8] recently reported that the Mercodia sandwich ELISA showed 17.3, 10.2, and 22.7% cross-reactivity with glicentin, oxyntomodulin, and glicentin1–61, respectively. Therefore, there is a definite need to develop a more accurate glucagon assay system and to evaluate its performance against currently available immunoassays.

Schematic of glucagon and related proglucagon products. The numbers of amino acid residues corresponding to proglucagon are shown at the top. Glucagon is shown in black, and peptides sharing the amino acid sequence with glucagon are shown in gray. GRPP glicentin-related pancreatic polypeptide, IP-1 intervening peptide-1, IP-2 intervening peptide-2
Owing to its high specificity, sensitivity, and relatively short developing time, there has been an increasing interest in the use of liquid chromatography-mass spectrometry (LC-MS) for peptide quantification. In addition, this method makes it possible to distinguish modified peptides, including oxidized peptides, from intact peptides. However, MS-based methods have not achieved the same widespread usage as immunoassay-based techniques, as they do not offer the same sensitivity. Although there have been several reports regarding the quantification of glucagon using LC-MS/MS [9], their sensitivities are not sufficiently high to quantify plasma levels during an oral glucose tolerance test (OGTT). Recently, Chappell and Lee et al. applied two antibodies to enrich for target peptides using LCMS/MS selected reaction monitoring (SRM): the first recognizes glucagon-like peptide-1 (GLP-1) and the second recognizes glucagon and oxyntomodulin [10, 11]. Using these antibodies, they developed an immunoprecipitation (IP)-LC-MS/MS SRM method to quantify four proglucagon-derived peptides (GLP-17–36 amide, GLP-19–36 amide, glucagon, and oxyntomodulin) simultaneously and reported that the lower limit of quantification (LLOQ) and total recovery rate for glucagon were 0.78 pM and 35.6%, respectively, using 500 μL of plasma [11]. Although the technique of pretreatment with IP is excellent, it is time-consuming in terms of the development of a suitable antibody for IP. Instead, SRM, which can be performed in a triple-quadrupole mass spectrometer, is currently the method of choice for the targeted quantification of peptides [12]. Additionally, several studies have suggested that parallel reaction monitoring (PRM), which can be performed with high-resolution mass spectrometry (HRMS), is more specific than SRM [13].
Here, we developed a novel method for measuring glucagon levels in human plasma using LC-HRMS. This LC-MS/MS PRM method was used to determine glucagon profiles during the OGTT and meal tolerance test (MTT) in healthy volunteers. Furthermore, the performance of this LC-MS/MS PRM method was evaluated in comparison to that of two commercially available immunoassays.
Materials and methods
Sample preparation for quantification of glucagon in human plasma
Preparation of plasma without glucagon (glucagon-free plasma)
Blood was collected in tubes containing lyophilized protease inhibitors, dipeptidyl peptidase-4 inhibitor, and EDTA-2K (all from Becton Dickinson and Company (BD P-800), Franklin Lakes, NJ, USA). To obtain glucagon-free plasma, 2 g of charcoal (charcoal activated A, powder; Nacalai Tesque, Inc., Kyoto, Japan) was added to every 10 mL of plasma, and this mixture was agitated for 12 h at 4 °C. After centrifuging for 1 h at 4 °C and 105,000×g, the supernatant was filtered using a 0.22-μm filter [14].
Protein precipitation and solid-phase extraction
To prepare a glucagon standard curve, a 200-μL charcoal-stripped plasma sample aliquot and 10 μL of a solution containing 200 μg/mL of sitagliptin (an inhibitor of dipeptidyl peptidase-4, synthesized by Sanwa Kagaku Kenkyusho, Mie, Japan) were mixed in a 2-mL tube. Glucagon (molecular mass 3482.7 Da, chemical purity ≥98%; Peptide Institute, Inc., Osaka, Japan) or isotopically labeled glucagon (14-Leu-[13C6]-glucagon, H-7236, molecular mass 3488.7 Da, chemical purity >95%, and 27-Met-sulfoxide-glucagon, H-6148, molecular mass 3498.8 Da, chemical purity >98%; Bachem, Bubendorf, Switzerland) was dissolved in 0.1% acetic acid (AcOH, LC-MS grade; Wako Pure Chemical Industries, Ltd., Osaka, Japan) to prepare a 100 μM standard solution. Both standards were diluted to 1 μM in 80% ethanol (EtOH, high-performance liquid chromatography (HPLC) grade; Wako) containing 0.1% AcOH, divided into aliquots, and stored at −30 °C. Then, aliquots of glucagon standard and internal standard (IS) solution were added to prepare a standard curve for glucagon ranging in concentration from 0.5 to 100 pM. The calibration curve for glucagon consisted of the following ten calibrators: blank, 0, 0.5, 1, 2, 5, 10, 20, 50, and 100 pM. For quantitative measurement of glucagon in plasma samples, 20 fmol of 14-Leu-[13C6]-glucagon was added as an IS into every 200 μL of glucagon standard or plasma sample. Subsequently, the samples were mixed with 100 μL of a 5% ammonia solution (NH4OH, special grade; Wako) and 900 μL of ice-cold EtOH, stored for 20 min at −40 °C, and centrifuged for 10 min at 20,400×g. Supernatants were collected into 15-mL tubes containing 1.5 mL of 5% NH4OH and were then loaded onto an Oasis MAX 96-well plate (30 μm particle size, 30 mg; Waters, Milford, MA, USA), which was equilibrated with 1 mL of methanol (MeOH, LC-MS grade; Wako) and 1 mL of water. The wells were subsequently washed with 5% NH4OH, 1 mL of 75% acetonitrile (ACN, LC-MS grade; Wako), 200 μL of water, and 1 mL of 10% AcOH. Finally, glucagon was eluted from the wells with 400 μL of 40% ACN containing 10% AcOH, and the eluents were evaporated to dryness. The dried residues were reconstituted with 30 μL of 50% ACN containing 1% AcOH and 5 mM l-methionine (special grade; Wako). Following filtration with a 0.20-μm filter, 5 μL of the extracted sample was used for analysis (see Electronic Supplementary Material (ESM) Fig. S1).
LC-MS/MS conditions
Quantification was performed on an automated nanoLC-MS/MS system consisting of an Ultimate 3000 Series nanoLC system and a Q Exactive quadrupole/Orbitrap mass spectrometer (Thermo Fisher Scientific, GmbH, Bremen, Germany) equipped with a nano-electrospray ionization (ESI) interface Black XYZ ion source (AMR, Inc., Tokyo, Japan). Instrument control, data acquisition, and processing were performed using the associated Xcalibur 3.0 software. Extracted samples were loaded onto the trapping column (Acclaim PepMap C18, 5 μm particle size, 300 μm i.d. × 1 mm; Thermo Fisher Scientific, Waltham, MA, USA) equilibrated with 10% ACN containing 0.1% trifluoroacetic acid (TFA, HPLC grade; Wako) and were separated by nanoLC with the analytical column (nano-HPLC capillary column C18, 3 μm particle size, 75 μm i.d. × 100 mm; Nikkyo Technos, Co., Ltd., Tokyo, Japan) for 48 min at a flow rate of 200 nL/min with a linear gradient elution of 30–95% MeOH containing 0.1% formic acid (FA, LC-MS grade; Wako). For re-equilibration, 30% MeOH containing 0.1% FA was passed through the system for 12 min. The ambient temperature of the analytical column and that of the autosampler were set at 50 and 10 °C, respectively. The analytical column Ultrafree centrifugal filters Durapore PVDF (0.22 μm) and Millex LG PTFE (0.20 μm) were purchased from Merck Millipore (Darmstadt, Germany).
The MS system was tuned to glucagon, which was infused directly, resulting in the following parameters: polarity positive, resolution of 70,000 at m/z 200, capillary temperature of 350 °C, and spray voltage of 1800 V. The lock mass function was used for the Orbitrap system to obtain constant mass accuracy during the gradient elution. The calibrator ions were the plasticizer contaminant N-butylbenzenesulfonamide (C10H15NO2S) and diethylhexyl phthalate (C24H38O4) found in the mobile phase at m/z 214.08963 and m/z 391.28429, respectively. In PRM mode, the precursor ions were isolated by the quadrupole with a 1.5-m/z window and trapped in the curved linear trap (C-trap) sequentially with a maximum injection time of 1500 ms, followed by fragmentation with a normalized collision energy of 20 arbitrary units in the HCD collision cell. The AGC target, which controls the number of ions into the Orbitrap, was 2 × 105. PRM was performed by monitoring the summed peak area of five fragment ions of the +4 charge state for glucagon (871.66), 14-Leu-[13C6]-glucagon (873.17), and oxidized glucagon: 27-Met-sulfoxide-glucagon (875.69). Fragment ions are summarized in Fig. 2.

Representative chromatograms of glucagon along with corresponding IS peaks extracted from blank, charcoal-stripped human plasma (a), plasma spiked with 0.5 pM glucagon (b), and healthy participant plasma (c). Representative chromatograms of oxidized glucagon (27-Met-sulfoxide-glucagon) along with glucagon extracted from healthy participant plasma (d). Arrows show the expected retention time of glucagon (a) and oxidized glucagon (d), respectively. Insets show peak intensity. Bottom insert parallel reaction monitoring transitions for glucagon peptides. The chromatograms are the reconstructed ion chromatograms of the sum of five fragment ions
To prepare the calibration curve, the peak area ratios of glucagon to the IS for each standard sample (ranging from 0.5 to 100 pM, 8 points) and the known concentrations of the standard samples were plotted on the y-axis and x-axis, respectively. Glucagon concentrations in plasma samples were calculated by linear regression with a 1/x 2 weighting factor and are expressed in terms of the concentrations of the synthetic glucagon standard in the present study (ESM Fig. S2).
Method validation
Recovery of glucagon after extraction
To determine the recovery rate of glucagon, three charcoal-stripped plasma samples were spiked with 100 pM of glucagon either prior to or after extraction. The corresponding IS was spiked post extraction. The recovery rates were calculated by comparing the mean peak area ratio of the analyte to that of the IS of the samples.
Accuracy, precision, and LLOQ
To evaluate the intra-day and inter-day accuracy and precision of the procedure, three validation batches were prepared on separate days. Each batch included a set of calibration standards and five replicates of samples spiked with lower limit and low, medium, and high levels of glucagon (0.5, 1, 20, and 80 pM, respectively). Intra-day assay accuracy and precision were calculated as the percent relative error (%RE) and percent coefficient of variation (%CV) of five replicates, respectively. Inter-day assay accuracy and precision were determined on three separate days. LLOQ was defined as the concentration that passed prespecified criteria (intra-day and inter-day assay CV and extrapolated concentration not exceeding 20% of the nominal value).
Long-term stability in plasma
To determine the long-term stabilities of glucagon in plasma collected using BD P-800 tubes, plasma samples spiked with 50 pM of glucagon were stored at −20 and −80 °C for 3 months. Measurements were performed using triplicate samples for each storage temperature.
Stability after extraction
To determine glucagon stability after extraction, extracts from healthy participants were stored at 10 °C for 7 days. The postulated oxidized glucagon was identified via PRM, as indicated in Fig. 2.
Plasma levels of glucagon in healthy participants
The present LC-MS/MS method was applied to the determination of the plasma glucagon levels of healthy volunteers during the OGTT and MTT, which were compared with the levels of glucagon measured by a commercially available sandwich ELISA kit, which uses monoclonal antibodies against both the C-terminal and N-terminal of glucagon (10-1271-01; Mercodia, Uppsala, Sweden) and an RIA kit, which uses polyclonal antibodies against the C-terminal of glucagon (GL-32K; Merck Millipore, MA, USA). Male volunteers were subjected to the OGTT or MTT in the morning after an overnight fast [n = 7, mean age = 35.1 (range 24–43) years, mean body mass index = 22.1 (20.1–25.3) kg/m2 for the OGTT; n = 13, mean age = 34.2 (range 24–52) years, mean body mass index = 22.4 (18.6–25.3) kg/m2 for the MTT]. Trelan-G75 (Yoshindo, Toyama, Japan; 75 g glucose) or Sanet-SA [Sanwa Kagaku Kenkyusho, Nagoya, Japan; 6 mL/kg body weight, maximum dose 360 mL (360 kcal), carbohydrate/protein/fat = 2.8:1:1] was ingested for the OGTT or MTT, respectively. Blood samples (approximately 2.5 mL) were collected into P-800 tubes at 0, 30, 60, 120, and 180 min. Plasma samples were stored at −80 °C until glucagon measurements.
Statistical analysis
Data are expressed as means ± SEM. For multiple comparisons of glucagon levels during the OGTT and MTT, repeated-measures one-way ANOVA was performed followed by a post hoc test with Bonferroni adjustment. Relationships between glucagon levels measured by different glucagon assays were assessed using Pearson’s correlation coefficient. Statistical analyses were performed using IBM SPSS Statistics, Ver. 22. P < 0.05 was considered statistically significant.
Results and discussion
There are two main methods for quantifying peptides by LC-MS. One is to quantify intact target peptides, and the other is to quantify a surrogate peptide, which is a fragment peptide derived from the target peptide. In general, it is possible to quantify peptides composed of about 40 amino acid residues using intact peptides. However, depending on the adsorption and solubility of the peptide, the method of quantification using the shorter surrogate peptide may be superior to that of quantification using the intact peptide in terms of the sensitivity of MS. The surrogate peptide should be unique and should be resistant to any modifications. In a previous report, we showed that the N-terminal peptide of GIP1–8/GIP3–8 obtained by digestion using endoproteinase Asp-N is a suitable surrogate peptide for intact GIP1–42/GIP3–42 in a simultaneous quantification method for GIP1–42 and GIP3–48 in human plasma. In this case, the surrogate peptide is not only more sensitive compared to the intact GIPs in LC-ESI-MS/MS analysis but also useful by a virtue of being difficult to adsorb [14]. Unlike GIP peptides, glucagon shares a common N-terminal end with oxyntomodulin (Fig. 1), and the metabolic processes of oxyntomodulin and glicentin are not clear. Therefore, enzymatic digestion cannot be used for a glucagon assay using LC-MS/MS. Thus, in the present LC-MS/MS study, we measured intact glucagon without using a peptidase pretreatment.
Assay validation
Specificity and recovery rate
Owing to the lack of mammalian plasma specimens that do not contain human glucagon, a test for specificity was carried out using glucagon-free plasma samples (n = 6). As revealed in Fig. 2a, no interfering signals were observed at the expected retention time for glucagon. The PRM chromatogram of the targeted transitions for glucagon spiked at 0.5 pM (equal to 16.7 amol on the column) and clearly displayed a peak corresponding to glucagon in the glucagon-free plasma (Fig. 2b). Furthermore, specificities were evaluated using plasma samples from healthy volunteers (Fig. 2c). Although plasma extracts treated with protein precipitation (PP) and solid-phase extraction (SPE) include a large number of peptides and small molecules, the PRM showed sufficient detection of glucagon. The total recovery rate of glucagon after PP and SPE was 82.1%.
The addition of isotopically labeled IS effectively overcomes the problems associated with fluctuations in signal intensity. Therefore, almost all peptide or protein quantification methods using LC-MS are performed with an isotope-labeled peptide as the IS [9,10,12, 14]. In the present LC-MS/MS method, 14-Leu-[13C6]-glucagon was added to every sample as the IS. Recoveries after PP and SPE were significantly corrected by this isotope-labeled full-length IS peptide, indicating that the method was highly effective as an accurate quantification method.
LC-HRMS has the versatile capability to perform reliable and sensitive quantitative and qualitative analyses or both (Quan/Qual) while recording in high-resolution full-scan or MS/MS (PRM) mode [13]. Several researchers have explored whether these assays can serve as alternatives to the conventional SRM mode using triple-quadrupole MS. Grund et al. [13] compared these different approaches using low molecular weight molecules. Although they did not reach a conclusion about a superior method, they indicated that the accuracy with respect to LOD levels was better for PRM mode than that obtained in full-scan mode in the quantification of testosterone in plasma. In addition, our preliminary quantification study of short peptides, consisting of eight amino acids, indicated that there was no background noise in PRM mode, whereas many intercept peaks were observed in SRM mode (data not shown). IP is an excellent technique to extract target peptides. However, the development of a suitable antibody for IP is time-consuming because of the need for antibody validation (e.g., cross-reactivity, recovery). Moreover, it is often difficult to achieve high recovery rates. Although we did not use IP methods to obtain the extract from plasma, the PRM approach and nanoLC made it possible to achieve high specificity and high sensitivity using only 200 μL of plasma.
Precision, accuracy, and LLOQ
Glucagon validation samples were analyzed at concentrations of 0.5, 1, 20, and 80 pM. These measurements showed accuracy within 12% and precision not exceeding 13% in intra-day assays, and accuracy within 4% and precision not exceeding 14% in inter-day assays (Table 1). Using this approach, LLOQ values of 0.5 pM and working ranges of 0.5–100 pM were obtained (ESM Fig. S2). This LLOQ is lower than that of the sandwich ELISA (1.5 pM) [15] and is also lower than that of IP-LC-MS/MS (0.78 pM requiring 500 μL of plasma) [11]. Accuracy and precision were as good as those achieved by IP-LC-MS/MS in SRM mode [11] when evaluating the glucagon profile.
Stability and oxidized glucagon
We predicted that glucagon would be oxidized because it contains methionine, which can be easily oxidized, at position 27 [16]. The oxidation of methionine should be taken into consideration in such analyses, since it will change the molecular weight of the peptide, which may lead to inaccurate glucagon measurements. Glucagon was stable in P-800 plasma tubes stored for 3 months at −20 and −80 °C (ESM Table S1). Oxidized glucagon was undetectable in the plasma of healthy volunteers (Fig. 3d). Glucagon oxidation was observed in plasma extracts after storage at 10 °C for 7 days without methionine but was only present at 0.2% relative to intact glucagon in samples stored with 5 mM methionine (ESM Fig. S3). Therefore, adding methionine to the final extraction solution is an effective method for assuring accurate glucagon level measurements in plasma.

Plasma glucagon responses in healthy volunteers quantified by the present LC-MS/MS method (a), sandwich ELISA (b), and RIA (c) during OGTT (solid circles, n = 7) and MTT (open circles, n = 13). Data represent the mean ± SEM. For better visualization of the data, significant differences from 0 min are indicated (*P < 0.05, **P < 0.01). The dotted line indicates the LLOQ of each assay. (a) 0.5 pM. (b) 1.5 pM. (c) 5.7 pM. The bottom two figures show the comparison of LC-MS/MS estimates of plasma glucagon levels in healthy volunteers with those of sandwich ELISA (d) or RIA (e)
Evaluation of plasma glucagon levels during the OGTT and MTT in healthy volunteers
The LC-MS/MS method was used to evaluate plasma glucagon levels during the OGTT and MTT in healthy volunteers, and its results were compared with values obtained by sandwich ELISA and RIA. As shown in Fig. 3a, according to LC-MS/MS, plasma glucagon levels significantly decreased during the OGTT in healthy volunteers, with a maximum decrease detected at 120 min. In contrast, plasma glucagon levels did not decrease but rather tended to increase, during the MTT. Importantly, as shown in Fig. 3b, similar patterns were observed for plasma glucagon levels measured by sandwich ELISA. However, plasma glucagon levels measured by RIA showed different patterns from those determined by LC-MS/MS; i.e., both the OGTT and MTT resulted in decreases in plasma glucagon levels (Fig. 3c). Although all samples showed higher glucagon values during the OGTT than the LLOQ of the LC-MS/MS method, 22.9% of the samples had glucagon values below the LLOQ of the sandwich ELISA (ESM Table S2), indicating that the LC-MS/MS method may be more sensitive at lower levels of glucagon than the sandwich ELISA.
Plasma glucagon levels have been reported to decrease after glucose loading in healthy humans [7, 8, 17]. In a previous study that measured glucagon levels during the MTT in healthy humans by microflow IP-LC-MS/MS, glucagon levels were significantly elevated after meal intake, with a peak at 90 min [11]. In contrast, in the present study, glucagon levels increased only moderately during the MTT until 180 min after ingestion. In the previous study, however, the participants received a test meal of one Nestlé Boost drink and one 30-g ProteinPlus PowerBar, which contains twice as much protein as that in the test meal given in the present study. These results suggest that protein stimulates while glucose suppresses the secretion of glucagon.
Comparison of LC-MS/MS with sandwich ELISA and RIA
Glucagon values were plotted to compare the performance of LC-MS/MS with that of commercially available immunoassays (Fig. 3d, e). Values of glucagon measured by sandwich ELISA were generally lower than those measured by the present LC-MS/MS method. Standards for glucagon immunoassays are calibrated against WHO 1st international reference preparation 69/194, which was originally extracted from porcine pancreases for the purpose of glucagon bioassays, not for immunoassays. This may result in different values of glucagon among assay kits. Nonetheless, the correlation between the glucagon values obtained by these two methods was much better (r = 0.879, Fig. 3d) than that between RIA and LC-MS/MS (r = 0.546, Fig. 3e, ESM Tables S2, S3). Despite the better correlation with sandwich ELISA, these values were still not perfectly matched, probably due to the limitations of the immunoassay-based method, i.e., antibody cross-reactivity with other peptides, which has been claimed by several groups [5,6,7,8].
Conclusions
In conclusion, we developed a novel method to measure glucagon by LC-MS/MS, which provides higher accuracy and precision for quantitative assessments of glucagon in human plasma than possible with currently available immunoassays. This LC-MS/MS method can be used for the analysis of plasma glucagon levels during the OGTT and MTT in diabetes patients as well as healthy individuals. The glucagon profile obtained by LC-MS/MS should contribute to gaining a better understanding of the physiological and pathological importance of glucagon.
Abbreviations
- CV:
-
Coefficient of variation
- ESI:
-
Electrospray ionization
- HRMS:
-
High-resolution mass spectrometry
- IS:
-
Internal standard
- LLOQ:
-
Lower limit of quantification
- MTT:
-
Meal tolerance test
- OGTT:
-
Oral glucose tolerance test
- PP:
-
Protein precipitation
- PRM:
-
Parallel reaction monitoring
- RE:
-
Relative error
- SPE:
-
Solid-phase extraction
References
Mighiu PI, Yue JTY, Filippi BM, Abraham MA, Chari M, Lam CK, et al. Hypothalamic glucagon signaling inhibits hepatic glucose production. Nat Med. 2013;19(6):766–72.
Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122(1):4–12.
Unger RH, Eisentraut AM, McCall MS, Keller S. Glucagon antibodies and their use for immunoassay for glucagon. Proc Soc Exp Biol Med. 1959;102:621–3.
Bataille D, Dalle S. The forgotten members of the glucagon family. Diabetes Res Clin Pract. 2014;106(1):1–10.
Bak MJ, Albrechtsen NW, Pedersen J, Hartmann B, Christensen M, Vilsbøll T, et al. Specificity and sensitivity of commercially available assays for glucagon and oxyntomodulin measurement in humans. Eur J Endocrinol. 2014;170(4):529–38.
Holst JJ, Christensen M, Lund A, de Heer J, Svendsen B, Kielgast U, et al. Regulation of glucagon secretion by incretins. Diabetes Obes Metab. 2011;13(Suppl 1):89–94.
Albrechtsen NJW, Hartmann B, Veedfald S, Windeløv JA, Plamboeck A, Bojsen-Møller KN, et al. Hyperglucagonaemia analysed by glucagon sandwich ELISA: nonspecific interference or truly elevated levels? Diabetologia. 2014;57(9):1919–26.
Matsuo T, Miyagawa J, Kusunoki Y, Miuchi M, Ikawa T, Akagami T, et al. Postabsorptive hyperglucagonemia in patients with type 2 diabetes mellitus analyzed with a novel enzyme-linked immunosorbent assay. J Diabetes Invest. 2016;7(3):324–31.
Chambers EE, Lame ME, Rainville PD, Murphy J, Johnson J, Fountain KJ, et al. Practical applications of integrated microfluidics for peptide quantification. Bioanalysis. 2015;7(7):857–67.
Chappell DL, Lee AYH, Castro-Perez J, Zhou H, Roddy TP, Lassman ME, et al. An ultrasensitive method for the quantitation of active and inactive GLP-1 in human plasma via immunoaffinity LC-MS/MS. Bioanalysis. 2014;6(1):33–42.
Lee AYH, Chappell DL, Bak MJ, Judo M, Liang L, Churakova T, et al. Multiplexed quantification of proglucagon-derived peptides by immunoaffinity enrichment and tandem mass spectrometry after a meal tolerance test. Clin Chem. 2016;62(1):227–35.
Sano S, Tagami S, Hashimoto Y, Yoshizawa-Kumagaye K, Tsunemi M, Okochi M, et al. Absolute quantitation of low abundance plasma APL1β peptides at sub-fmol/mL level by SRM/MRM without immunoaffinity enrichment. J Proteome Res. 2014;13(2):1012–20.
Grund B, Marvin L, Rochat B. Quantitative performance of a quadrupole-orbitrap-MS in targeted LC-MS determinations of small molecules. J Pharm Biomed Anal. 2016;124:48–56.
Miyachi A, Murase T, Yamada Y, Osonoi T, Harada K. Quantitative analytical method for determining the levels of gastric inhibitory polypeptides GIP1–42 and GIP3–42 in human plasma using LC-MS/MS/MS. J Proteome Res. 2013;12(6):2690–9.
Auld CA. White paper “Glucagon measurement—addressing long-standing analytical challenges.” 2015. https://www.mercodia.se. Accessed 24 Oct 2016.
Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4(222):1–14.
Bagger JI, Knop FK, Lund A, Holst JJ, Vilsbøll T. Glucagon responses to increasing oral loads of glucose and corresponding isoglycaemic intravenous glucose infusions in patients with type 2 diabetes and healthy individuals. Diabetologia. 2014;57(8):1720–5.
Acknowledgements
We are grateful to the members of Thermo Fisher Scientific and to T. Murase, S. Yamashita, and H. Hashimoto for their fruitful discussions and technical support. We also thank the members of the Kitamura Laboratory for the discussion of the data.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All participants gave oral and written consent. The study was approved by the Ethics Committee of Gunma University and was conducted in accordance with the Declaration of Helsinki.
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(PDF 261 kb)
Rights and permissions
About this article

Cite this article
Miyachi, A., Kobayashi, M., Mieno, E. et al. Accurate analytical method for human plasma glucagon levels using liquid chromatography-high resolution mass spectrometry: comparison with commercially available immunoassays. Anal Bioanal Chem 409, 5911–5918 (2017). https://doi.org/10.1007/s00216-017-0534-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0534-0