
Abstract
Benzothiazoles are high production volume chemicals widely used in many industrial and household applications. However, information on their occurrence in aquatic organisms is very limited, although a high level of bioaccumulation is expected. In this study and for the first time, a method was developed involving subcritical water extraction followed by solid-phase microextraction coupled to gas chromatography-ion trap-tandem mass spectrometry for the determination of five benzothiazoles in seafood. The repeatability and reproducibility of the method were under 21% (%RSD, n = 5, 100 ng g−1 (dw)), while method detection limits and method quantification limits were between 0.5 and 10 ng g−1 (dw) and 1 and 50 ng g−1 (dw), respectively. Ten widely consumed fish and shellfish species from the county of Tarragona (Catalonia, Spain) were selected in order to estimate dietary exposure and to assess the human health risks. The most frequently determined compounds were benzothiazole and 2-(methylthio)-benzothiazole, with squid being the species which showed the highest level of benzothiazole (82 ng g−1 (dw)). In terms of human exposure, the current concentrations of benzothiazoles found in fish and shellfish could not be compared to threshold values because of the lack of toxicological data.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Benzothiazoles include a large class of high production volume chemicals with a very broad range of applications in industry and household products [1]. Due to the large amount of several chemicals currently used, high production may indicate high exposure, with inherent risks for health and the environment. Benzothiazoles are commercially manufactured for use as vulcanization accelerators in rubber production. In addition, they are used as biocides in paper and leather manufacture, corrosion inhibitors in antifreeze formulations [2], fungicides and herbicides [3], and ultraviolet light stabilizers in textiles and plastics [4]. Moreover, benzothiazoles comprise a class of therapeutic compounds that display anticancer [5], antimicrobial, antiviral and antidiabetic activity, among other benefits [6]. Their structure consists of a five-membered 1,3-thiazole ring fused to a benzene ring. The nine atoms of the bicycle and the attached substituents are coplanar. Furthermore, they are structurally related to naturally occurring purines due to their interaction with charged biomolecules. This structural concordance may pose a human risk because there might be interactions between benzothiazoles and some proteins [5, 7].
Benzothiazole (BT) is recognized as a safe substance, being used as a flavouring in food applications [8]. Despite the lack of toxicological studies [9], the European Food Safety Authority (EFSA) has set a limit in food of 0.05 mg kg−1. However, some benzothiazoles have shown toxic effects in in vivo tests with fish cell cultures, including cell death but at concentrations higher than those reported in environmental samples [10]. Ginsberg et al. [9] indicated that BT may pose a high risk at sufficient exposure, exhibiting adverse effects on the liver and kidney, as well as dermatitis and respiratory problems [7, 9, 11]. However, the available information on the aquatic toxicology of benzothiazoles, and especially their effect on fish, is still very limited. Therefore, the knowledge of human risks due to exposure to benzothiazoles through seafood consumption is scarce. According to risk assessment data from the World Health Organization (WHO), an oral non-observed adverse effect level (NOAEL) of 5.1 mg kg bw−1 day−1 has been set for BT, based on a 90-day dietary study on rats [12]. However, no threshold values have yet been established for other benzothiazoles.
Since benzothiazoles are easily released into the environment, it is not surprising that these compounds have been detected in a wide variety of matrices, such as wastewater [13,14,15,16,17], river water [2], sewage sludge [16,17,18,19], human urine [7, 20], adipose tissue [21], house dust and indoor air [22], clothing textiles [4] and synthetic turf [9], among others. As a consequence of their presence in river water and sewage sludge, trace amounts of these compounds may be expected in aquatic organisms. Unfortunately, to the best of our knowledge, analytical methods to determine benzothiazoles in aquatic organisms have not been developed and validated yet.
Nevertheless, there are a few methods that have been developed to determine benzothiazoles in solid samples which use extraction techniques such as pressurized liquid extraction (PLE) [18], ultrasound-assisted solvent extraction (UASE) [4] and liquid-solid extraction (LSE) based on quick, easy, cheap, effective, rugged and safe (QuEChERS) extraction [19]. Furthermore, sample pretreatment is necessary before extraction, which usually includes a freeze-dry step to avoid the presence of water that could distort the results. The matrix effect becomes critical when complex matrices are analysed. The source of matrix effect in biological samples usually arises from the free fatty acids responsible for inducing an enhancement or suppression of the signal, or even changing the retention time. Hence, clean-up steps are necessary to improve the identification and quantification of target compounds. Moreover, some research on aquatic organisms has shown a significant positive correlation between the accumulation of chemicals and the lipid content of organisms [23]. For this reason, lipid determination in fish samples is commonly used in order to assess the bioaccumulation of the analytes.
The most widely used technique for the analysis of benzothiazoles is liquid chromatography (LC) and, to a lesser extent, gas chromatography (GC), both coupled to mass spectrometry (MS) or tandem mass spectrometry (MS/MS) [2, 4, 14, 16, 20]. Despite the matrix effect being a major problem, electrospray ionization (ESI) is preferred when working with LC. To overcome this disadvantage, atmospheric pressure chemical ionization (APCI) is sometimes used as an alternative, since it is less vulnerable to matrix effects [17, 24, 25]. In contrast, the interaction of analytes and matrix components during sample preparation is the main matrix impact in the case of GC, rather than ionization [26]. In any case, little is known about analytical methods combining the determination of benzothiazoles by GC in solid samples, whether biological or otherwise [25]. Moreover, most of the few studies on benzothiazole analysis by means of GC focus on liquid samples, such as water or urine [14, 20, 27].
The main goal of this study was to develop, for the very first time, a method based on gas chromatography-ion trap-tandem mass spectrometry (GC-IT-MS/MS) to determine five benzothiazoles in ten species of seafood. Subcritical water extraction (SBWE) and solid-phase microextraction (SPME) were used to extract and preconcentrate the analytes. Concentration data were used to assess the dietary exposure to those compounds through seafood consumption and to characterize the human health risks for the consumers in the county of Tarragona (Spain).
Experimental
Reagents and standards
The target benzothiazoles (benzothiazole (BT), 2-chlorobenzothiazole (ClBT), 2-aminobenzothiazole (NH2BT), 2-hydroxybenzothiazole (OHBT) and 2-(methylthio)-benzothiazole (MeSBT)) were purchased from Sigma-Aldrich (St. Louis, USA). Individual stock solutions were prepared in methanol at 1000 mg L−1 and stored at −20 °C. A working mixture solution was also prepared in methanol at 10 mg L−1 and stored in a refrigerator until use.
Methanol, hexane and dichloromethane were GC grade with purity >99.9% from J.T. Baker (Deventer, the Netherlands). Ultrapure water was obtained using an ultrapure water purification system from Veolia Water (Sant Cugat del Vallès, Barcelona, Spain). Helium gas with a purity of 99.999% was used for the chromatographic analysis (Abelló Linde, Barcelona, Spain).
Sample preparation
Seafood was obtained from three commercial establishments (supermarket, fish store and local market) in the county of Tarragona (Spain) to ensure samples from a different origin. Samples of cod (Gadus morhua), salmon (Salmo salar), sole (Solea solea), mackerel (Scomber scombrus), mussel (Mytilus galloprovincialis), hake (Merluccius merluccius), sardine (Sardina pilchardus), tuna (Thunnus thynnus), shrimp (Aristeus antennatus) and squid (Loligo vulgaris) were selected as representing the most widely consumed species in Catalonia (Spain) [28]. The edible part of the seafood species was removed and then preserved in a refrigerator until use. Frozen homogenized samples were freeze-dried using the freeze-drying system (Labconco, Kansas City, MO, USA) and crushed using a mortar and pestle. Additionally, mussels were sieved through a 125-μm screen to homogenize the diameter of the particles. Lastly, a composite sample for each species was obtained by mixing equal amounts from the three different commercial establishments.
Spiked samples were prepared by adding the stock mixture of standards in acetone at the volume required to cover the freeze-dried fish sample. After spiking, the samples were stirred intensively so that there would be sufficient contact between the compounds and the matrix. The acetone was left to evaporate at room temperature in a fume cupboard with frequent homogenization of the sample.
Extraction procedure
Subcritical water extraction was carried out using an ASE 350 accelerated solvent extraction system (Dionex, Sunnyvale, CA, USA) with an 11-mL stainless steel extraction cell. A glass fibre filter was placed at the bottom of the cell; then, 1 g of diatomaceous earth (Thermo Scientific, Barcelona, Spain) was added, followed by 1 g of freeze-dried sample previously mixed with 1 g of diatomaceous earth and then 1 g of diatomaceous earth, which filled up the cell. Prior to extraction, on-cell clean-up was performed with hexane to remove lipids following conditions adapted from a previous study [18]: temperature at 60 °C, two cycles of 5 min each, 5 min static time, 1500 psi, flush volume of 80% and a purge time of 300 s. Then, the extraction was carried out with one cycle of 5 min using ultrapure water as solvent extraction at 80 °C and 1500 psi. The preheating time was 5 min, the flush volume was 60% of the cell volume and the purge time was 120 s. After extraction, 10 mL of the PLE extract was taken to perform the SPME.
The suitability of two SPME fibres (PA 85 μm and PDMS/DVB 65 μm, both from Supelco (Bellefonte, PA, USA)) was checked, with PDMS/DVB 65 μm selected to conduct the SPME extraction. The fibre was conditioned in line with the supplier’s instructions, being inserted into the GC injector. Ten millilitres of SBWE aqueous extract was poured into a 20-mL SPME vial and immediately sealed tight with a Teflon septum and placed in the tray of the CombiPAL autosampler (CTC Analytics, Zwingen, Switzerland), which allowed full automation of the SPME. After an equilibration time of 5 min, the PDMS/DVB 65-μm fibre was immersed in the water solution for 40 min at 80 °C. During the extraction, the sample was magnetically stirred at 750 rpm. Afterwards, desorption took place at the GC injector at 270 °C for 3 min, and the compounds were subsequently determined by GC-IT-MS/MS.
To prevent carry-over, the PDMS/DVB 65-μm fibre was cleaned by heating at 270 °C for 10 min prior to every extraction and a blank test was performed to check for possible carry-over.
Chromatographic analysis
The GC-IT-MS/MS analyses were performed on a Varian 3800 gas chromatograph (Varian, Walnut Creek, CA, USA) connected to a Varian 4000 ion trap mass detector. The GC was equipped with a 1079 programmable temperature vaporizing injector and a 0.8-mm i.d. insert liner (Varian). A ZB-5 Plus analytical column (30 m × 0.25 mm i.d., 0.25 μm film thickness) from Phenomenex (Torrance, CA, USA) was used for the chromatographic separation. Varian MS Workstation v.6.9 software was used for instrument control and data processing.
For the chromatographic analysis, the injector was operated in splitless mode at 270 °C. The oven temperature was programmed as follows: 85 °C held for 3.5 min, raised at 25 °C min−1 to 200 °C, then 5 °C min−1 to 250 °C and finally 10 °C min−1 to 280 °C and held for 2 min. The carrier gas employed was helium at a constant flow rate of 1 mL min−1. The target compounds were separated in 10 min. Transfer line, manifold and trap temperatures were 280, 50 and 200 °C, respectively. The mass spectrometer was operated in the electron ionization (EI) mode at 70 eV. For quantitative analysis of the target compounds, tandem mass spectrometry (MS/MS) mode was applied. Table 1 summarizes the retention time and the optimal MS conditions for each compound.
Exposure assessment and risk characterization
The dietary exposure assessment combines food consumption data with the concentration of chemicals detected in food. As such, the resulting dietary exposure may be compared with values from databases on chemicals in food. The formula used to calculate the dietary exposure is as follows:
where E t is the global dietary exposure to the benzothiazole t in the general population (ng kg bw−1 day−1), C f is the mean consumption of the seafood species f by population (g kg bw−1 day−1) and X t, f represents the concentration of benzothiazole t in the seafood species f (ng g−1). The mean consumption was previously normalized, dividing the dietary intake by the mean body weight (bw). Contamination data was implemented on a wet weight (ww) basis. The concentration of non-detected analytes and analytes below method quantification limits (MQL) was set as the upper-bound (UB) intake, where the concentration of non-detected analytes was assumed to be the corresponding method detection limit (MDL) values and the concentration of analytes below MQL was assumed to be their MQL values [29, 30]. Therefore, the dietary exposure was reported using the sum of exposure to benzothiazoles through seafood consumption, taking into account different subpopulation groups, classified according to age (10–19, 20–65 and 65–80 years old) and gender.
The dietary risk was characterized by dividing the global dietary exposure by the provisional tolerable daily intake, estimated from the oral NOAEL, and considering an uncertainty factor of 1000 [31], as a margin of safety. The equation used to characterize the risk is as follows:
where R t is the health risk due to the dietary exposure to the benzothiazole t, E t is the dietary exposure to the benzothiazole t (ng kg bw−1 day−1) and pTDI t is the provisional tolerable daily intake for the benzothiazole t (ng kg bw−1 day−1).
Consumption data were collected from the Nutritional Survey of Catalonia [28], performed in 2002–2003 with 2160 individuals (54% female and 46% male). The volunteer participants were aged between 10 and 80, representing the whole Catalonia population. The consumption data was provided by means of 3-day dietary records, 24-h records and a food frequency questionnaire. The consumption rates of the general population of all the analysed species (hake, cod, sole, squid, shrimp, mussel, tuna, mackerel, salmon and sardine) are summarized in Table 2.
Results and discussion
Method optimization
GC-IT-MS/MS optimization
A mixed solution of 1 μg L−1 of the five benzothiazoles was prepared in methanol. One microlitre of this solution was injected directly into the GC-IT-MS/MS system, under full-scan acquisition mode. The target compounds were identified by their molecular ion, and then, the chromatographic separation was optimized. As mentioned above, the separation was performed in 10 min. Although MeSBT, NH2BT and OHBT coeluted, they could be quantified separately, due to the differences in their molecular and fragment ions. To achieve maximum sensitivity/selectivity, MS/MS was carried out selecting appropriated precursor/product ions and then optimizing the ion trap MS/MS parameters described in Table 1.
Extraction and clean-up optimization
Because low concentrations of benzothiazoles in seafood samples were expected, the SBWE extracts were preconcentrated by SPME. Therefore, SPME optimization was performed first and SBWE parameters were subsequently adjusted.
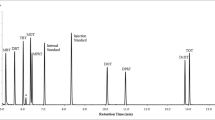
Firstly, the extraction efficiency of two fibres (PA 85 μm and PDMS-DVB 65 μm) was evaluated by comparison of the peak areas obtained in immersion mode under the working conditions described by Naccarato et al. [20], who determined a larger group of contaminants in aqueous matrices, including some of the same benzothiazoles here analysed. Then, 10 mL of ultrapure water spiked at 1 mg L−1 was extracted at 30 °C for 40 min. Desorption was carried out for 3 min at 270 and 290 °C for PDMS/DVB 65 μm and PA 85 μm fibres, respectively. Because higher areas for almost all compounds were obtained with PDMS-DVB 65 μm (Fig. 1), this fibre was chosen to perform the SPME. Once the fibre was selected, various extraction temperatures (30, 50, 70 and 80 °C) were tested in order to increase peak areas without sacrificing analysis time. The peak areas of BT, ClBT and MeSBT increased by 90% at 80 °C. However, NH2BT and OHBT showed no significant differences between all of the tested temperatures. Therefore, 80 °C was selected as the optimal SPME extraction temperature. The other extraction parameters were kept at initial values because areas did not improve by varying them.

Comparison of the chromatographic peak areas obtained with PDMS-DVB 65-μm and PA 85-μm SPME fibres under the same conditions. For other conditions, see text
Then, SBWE was optimized and initial conditions were adapted from a previous study [18] in which a group of benzothiazoles, benzotriazoles and benzenesulphonamides were determined in sludge. The conditions were as follows: one cycle of 5 min, ultrapure water at 80 °C, 1500 psi, preheating time of 5 min, flush volume of 60% and purge time of 120 s. Although there are several parameters of PLE extraction to optimize, solvent extraction, temperature and extraction time have the largest influence. In this case, only the extraction time was optimized because water was needed as the extraction solvent to facilitate the SPME step and the initial temperature was high enough with respect to the extraction solvent used. Therefore, 5 and 10 min were tested using 1 g of freeze-dried hake sample spiked at a concentration of 10 mg kg−1 (dw). Benzothiazole was the compound that showed the highest differences between the two times tested, with a peak area 40% higher at 5 min. ClBT and OHBT also showed slightly higher peak areas at 5 min, while NH2BT and MeSBT did not display any significant change. Thus, the extraction time selected was 5 min, as in the initial conditions.
Working with seafood samples, clean-up strategies are usually applied in order to remove interfering substances such as fat and oil, which may hinder the determination of trace levels [23]. In this study, on-cell clean-up and liquid-liquid extraction (LLE) were tested.
One gram of freeze-dried hake sample spiked at 10 mg kg−1 (dw) was used to test the different clean-up strategies. The on-cell clean-up was adapted from Hoff et al. [32], with the following conditions: hexane as the solvent extraction, temperature of 60 °C, two cycles of 5 min each one, 5 min static time, 1500 psi, flush volume of 80% and purge time of 300 s. In the case of liquid-liquid extraction clean-up, it was performed with the SBWE extract which was poured into the separating funnel and then 5 mL of hexane was added. The process was repeated five times with fresh hexane, which allowed a cleaner aqueous solution to be achieved. Because an emulsion between organic and aqueous phases was observed, 3 g of sodium chloride was added.
Comparing all of the clean-up results, benzothiazoles showed peak areas six times higher with on-cell clean-up than with LLE. Furthermore, the addition of salt in LLE neither improved the extraction nor eliminated the emulsion. Moreover, the on-cell clean-up allowed peak areas between 80 and 95% times higher than those obtained when no clean-up was used, with BT, ClBT and MeSBT being the compounds that showed a larger increase. Since on-cell clean-up seemed to be a good clean-up strategy, the matrix effect (ME) was determined by analysing two representative species of low (hake, 2%) and high (salmon, 25%) lipid content, which was determined using 1.5 g of lyophilized sample by PLE extraction with hexane:dichlorometane (1:1) followed by gravimetric determination [33]. The ME was calculated according to the following equation:
where B is the peak area of the analyte from a hake or salmon sample spiked after SBWE extraction, n is the peak area of the analyte present in the blank sample and A is the peak area of the analyte from the standard solution directly injected into the chromatographic system. As such, ME < 0 indicates ion suppression, while ME > 0 indicates ion enhancement. ME = 0 indicates that there is no matrix effect. When the on-cell clean-up strategy was applied, the ME was reduced, obtaining a range between −29 and 60% for hake and between −52 and −20% for salmon, in comparison to the results obtained without a clean-up step (from −54 to 100% and from −99 to 120%, respectively). Thus, on-cell clean-up with the aforementioned conditions was applied.
Method validation
Before validating the method, the matrix effect was studied by statistically comparing the slopes of the calibration curves (α = 0.05) for hake (low lipid content) and salmon (high lipid content). As expected, the matrix effect was observed for almost all of the compounds. Thus, both hake and salmon were used to validate the method as representative species of low and high lipid content, respectively. The method was validated by calculating the linear ranges, method detection limits (MDLs), method quantification limits (MQLs), apparent recoveries (R ap) and intra-day and inter-day repeatabilities (Table 3).
The linear range was evaluated by matrix-matched calibration by spiking hake and salmon at different concentrations. Non-spiked samples were also analysed to subtract the signal of the analytes present in the samples. The hake blank samples, corresponding to the white fish group, showed the presence of almost all the analytes. In turn, the salmon blank samples, corresponding to the fatty fish group, only showed the presence of BT and ClBT. To perform the matrix-matched calibration curves, eight calibration points for both groups of seafood samples were used and good linearity for all compounds was achieved (R 2 > 0.998) (Table 3).
MDLs corresponded to the concentration that caused a peak with a signal/noise ratio equal to 3 for the compounds that did not appear in the blanks. For the compounds present in the samples, they were estimated as the concentration that gave a signal average of three times higher than the standard deviation of the blank signal. Thus, MDLs were between 1 and 10 ng g−1 (dw) for hake and between 0.5 and 10 ng g−1 (dw) for salmon. MQLs were defined as the lowest points of the calibration curves, ranging from 5 ng g−1 (dw) to 50 ng g−1 (dw) for both seafood species. MDL and MQL values were consistent with those found in the literature for other solid environmental matrices, as there are no studies on the selected analytes in fish and shellfish. Stasinakis et al. [16] reported detection limits between 0.042 and 13 ng g−1 (dw) and quantification limits between 0.14 and 41 ng g−1 (dw) in the determination of BT, OHBT, MeSBT and NH2BT in sludge by solid-liquid extraction (SLE) followed by solid-phase extraction (SPE) and liquid chromatography. In another recent study, detection and quantification limits from 0.25 to 25 ng g−1 (dw) and from 0.5 to 50 ng g−1 (dw) were recorded when working with SBWE and SPE extractions, respectively, and liquid chromatography, for the determination of BT, OHBT, MeSBT and NH2BT in sludge [18].
Intra-day and inter-day repeatabilities (n = 5), expressed as relative standard deviation (%RSD), were calculated using 1 g of spiked sample of each matrix (n = 5) at concentrations of 50 and 100 ng g−1 (dw), and since the obtained results were comparable, Table 3 shows the values obtained at 50 ng g−1 (dw). They were lower than 21% for all of the compounds in each matrix. Apparent recoveries (R ap) were obtained from 1 g of spiked sample of each matrix (n = 5) at a concentration of 100 ng g−1 (dw) (Table 3). R ap were calculated using the following formula:
where A is the peak area of the analyte from hake or salmon spiked before SBWE extraction, n is the peak area of the analyte present in the blank sample and B is the peak area of the analyte from standard solution directly injected into the chromatographic system.
They ranged from 97 to 132% and 86 to 135% for hake and salmon, respectively. As expected based on their lipid content, when comparing both matrices, higher R ap values were reported for hake than for salmon.
Application to seafood samples
Benzothiazole levels in commercial seafood
The SBWE SPME GC-IT-MS/MS method was applied to determine benzothiazoles in ten species of fish and shellfish highly consumed in the county of Tarragona. Composite samples for each species were used in order to avoid origin influences. Furthermore, species were classified into two different groups (white fish and fatty fish) according to their lipid content (Table 4). To ensure the correct identification of the analytes, some criteria were taken into account. The retention time had to match that of the standard analyte within ±1 s, the signal-to-noise ratio (S/N) had to be ≥3 and deviation of the two monitoring ion intensities ratio had to be within 15% of that of the standard analyte. As already described, two matrix-matched calibration curves were used to quantify white and fatty fish species. The average concentrations (n = 5) of benzothiazoles in these ten commercial seafood species, expressed as dry weight (dw), are summarized in Table 4.
BT was found in almost all of the samples, being the compound with the highest concentration. Squid was the species with the greatest value (82 ng g−1 (dw)), while the minimum BT level corresponded to tuna (n.d.). The second most abundant analyte in seafood was MeSBT, the concentration of which ranged between n.d. and 24 ng g−1 (dw), for salmon and mussel, respectively. Unlike NH2BT, ClBT was more frequent in fatty fish samples than in white fish. Finally, OHBT was only found in two samples of white fish, hake and mussel, at concentrations <MQL. Mussel was the only species in which all benzothiazoles could be detected. The concentrations in mussels ranged between <MQL, for OHBT, and 58 ng g−1 (dw), for BT. Hake, squid and sardine showed four of the five analysed benzothiazoles.
To the best of our knowledge, this is the first study reporting the presence of benzothiazoles in seafood. Consequently, our results cannot be compared with those from previous studies. However, data seem to be in accordance with those already published related to the presence of benzothiazoles in water. For instance, a review focusing on the occurrence of benzothiazoles, among other contaminants, in the environment [25], highlighted the presence of BT, OHBT, MeSBT and NH2BT in effluent sewage water of different European countries and China, at low microgram per litre levels. Consequently, they can be present in superficial water and accumulate in biota.
Estimated dietary intake and risk evaluation
A summary of the estimated dietary exposure (ng kg bw−1 day−1) to BT, ClBT, MeSBT, NH2BT and OHBT through fish and shellfish consumption by the general population of Tarragona is depicted in Fig. 2. Human exposure was assessed for six population subgroups, based on age and gender, and assuming an upper-bound (UB) intake. Concentrations of benzothiazoles in seafood were recalculated and expressed on a wet weight (ww) basis for the human exposure assessment [34].

Estimated dietary intake (ng kg bw−1 day−1) of benzothiazoles for the general population of the county of Tarragona (Spain), classified by gender and age
Although BT was the most frequent compound, the highest estimated intake corresponded to MeSBT (22 ng kg bw−1 day−1) for senior women. It was followed by BT (11 ng kg bw−1 day−1) in adult women. Overall, these two compounds were the main contributors to the total intake of benzothiazoles. Women, both senior and adult, showed the greatest intake of all benzothiazoles, which was estimated at 48 ng kg bw−1 day−1. A linear increase of dietary intake with the age of the population was found (Fig. 2), with women displaying higher exposure than men.
Due to the very limited data concerning the oral safety levels of benzothiazoles, the risk evaluation was only attainable for BT. A pTDI was used, estimated from an oral NOAEL of 5.1 mg kg bw−1 day−1 [12]. This value was divided by 1000 as an uncertainty factor [31]. Thus, the pTDI used in the present study was 5100 ng kg bw−1 day−1. In a worst case scenario (UB intake), the risk was determined at 0.16 and 0.18% for boys and girls, 0.16 and 0.22% for adult men and adult women and 0.19 and 0.16% for senior men and senior women, respectively. As there is no threshold value for risk characterization, these values could not be compared.
Recent literature shows a few studies about human exposure to benzothiazoles, with a lack of information regarding risk assessment. One of them studied dust ingestion as a route of human exposure in the USA and East Asian countries. The results were categorized by age, reporting values of 2.871 and 0.452 ng kg bw−1 day−1 in children and adults in the USA and values ranging from 0.520 to 4.221 ng kg bw−1 day−1 and 0.104 to 0.911 ng kg bw−1 day−1 in children and adults from Asian countries [35]. Overall, those intake values were one to two orders of magnitude lower than the values reported in the present study, thus highlighting the potential role of food as an exposure pathway for benzothiazoles. In turn, other investigations have focused on non-dietary sources. The inhalation pathway was studied by Wan et al. [22], who reported a 95th percentile exposure of 9.24 and 6.86 ng kg bw−1 day−1 in two population groups of different ages: 12–21 years old and ≥21 years old, respectively. These data would be in line with values of the present research. In any case, more information is needed to identify the contribution of dietary routes and non-dietary pathways for exposure to benzothiazoles.
Conclusions
An analytical method based on SBWE SPME followed by GC-IT-MS/MS has been developed, for the first time, to enable the determination of benzothiazoles in complex matrices, such as seafood, at nanogram per gram (dw) levels. The method also provided good linearity, intra- and inter-day repeatability and MDLs and MQLs at low nanogram per gram levels.
To the best of our knowledge, this is the first study that demonstrates the presence of five benzothiazoles in ten commercial species of seafood, purchased in the county of Tarragona. Diet, and more specifically seafood consumption, was identified as a key pathway for exposure to benzothiazoles, although information on the contribution of other routes is still very limited. Our results showed detectable levels of BT and MeSBT in almost all of the samples, with squid being the species with the highest level of BT (82 ng g−1 (dw)). Moreover, mussel was the species which presented detectable amounts of all the benzothiazoles. Overall, the current intake of benzothiazoles through seafood consumption cannot be compared to threshold values because of the lack of reliable toxicological data.
References
OECD. (Organisation of Economic Co-operation and Development). Screening information data set for high production volume chemicals, UNEP Chemicals. 1998. http://www.oecd.org/chemicalsafety/risk-assessment/33883530.pdf. Accessed 14 Dec 2016.
Herrero P, Borrull F, Pocurull E, Marcé RM. Efficient tandem solid-phase extraction and liquid chromatography-triple quadrupole mass spectrometry method to determine polar benzotriazole, benzothiazole and benzenesulfonamide contaminants in environmental water samples. J Chromatogr A. 2013;1309:22–32.
He G, Zhao B, Denison MS. Identification of benzothiazole derivatives and polycyclic aromatic hydrocarbons as aryl hydrocarbon receptor agonists present in tire extracts. Environ Toxicol Chem. 2011;30:1915–25.
Avagyan R, Luongo G, Thorsen G, Ostman C. Benzothiazole, benzotriazole, and their derivates in clothing textiles. A potential source of environmental pollutants and human exposure. Environ Sci Pollut Res Int. 2015;22:5842–9.
Hassan N, Verdes PV, Ruso JM. Assessment of interactions between four proteins and benzothiazole derivatives by DSC and CD. J Chem Thermodyn. 2011;43:399–404.
Keri RS, Patil MR, Patil SA, Budagumpi S. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur J Med Chem. 2015;89:207–51.
Asimakopoulos AG, Wang L, Thomaidis NS, Kannan K. Benzotriazoles and benzothiazoles in human urine from several countries: a perspective on occurrence, biotransformation, and human exposure. Environ Int. 2013;59:274–81.
EFSA, (European Food Safety Authority). Safety and efficacy of thiazoles, thiophene and thiazoline belonging to chemical group 29 when used as flavourings for all animal species. 2016; doi: 10.2903/j.efsa.2016.4441.
Ginsberg G, Toal B, Kurland T. Benzothiazole toxicity assessment in support of synthetic turf field human health risk assessment. J Toxicol Environ Health A. 2011;74:1175–83.
Zeng F, Sherry JP, Bols NC. Evaluating the toxic potential of benzothiazoles with the rainbow trout cell lines, RTgill-W1 and RTL-W1. Chemosphere. 2016;155:308–18.
Ye Y, Weiwei J, Na L, Mei M, Kaifeng R, Zijian W. Application of the SOS/umu test and high-content in vitro micronucleus test to determine genotoxicity and cytotoxicity of nine benzothiazoles. J Appl Toxicol. 2014;34:1400–8.
Abbott PJ, Renwick AG. WHO Food Additives Series: 50. Sulfur-containing heterocyclic compounds. 1998. http://www.inchem.org/documents/jecfa/jecmono/v50je12.htm#1.0. Accessed 15 Dec 2016.
Asimakopoulos AG, Ajibola A, Kannan K, Thomaidis NS. Occurrence and removal efficiencies of benzotriazoles and benzothiazoles in a wastewater treatment plant in Greece. Sci Total Environ. 2013;452–453:163–71.
Fries E. Determination of benzothiazole in untreated wastewater using polar-phase stir bar sorptive extraction and gas chromatography-mass spectrometry. Anal Chim Acta. 2011;689:65–8.
Reemtsma T, Fiehn O, Kalnowski G, Jekel M. Microbial transformation and biological effects of fungicide-derived benzothiazoles determined in industrial wastewater. Environ. Sci. Technol. 1995;29:478–85.
Stasinakis AS, Thomaidis NS, Arvaniti OS, Asimakopoulos AG, Samaras VG, Ajibola A, Mamais D, Lekkas TD. Contribution of primary and secondary treatment on the removal of benzothiazoles, benzotriazoles, endocrine disruptors, pharmaceuticals and perfluorinated compounds in a sewage treatment plant. Sci Total Env. 2013;463–464:1067–75.
Wick A, Fink G, Ternes TA. Comparison of electrospray ionization and atmospheric pressure chemical ionization for multi-residue analysis of biocides, UV-filters and benzothiazoles in aqueous matrices and activated sludge by liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2010;1217:2088–103.
Herrero P, Borrull F, Marcé RM, Pocurull E. A pressurised hot water extraction and liquid chromatography-high resolution mass spectrometry method to determine polar benzotriazole, benzothiazole and benzenesulfonamide derivates in sewage sludge. J Chromatogr A. 2014;1355:53–60.
Herrero P, Borrull F, Pocurull E, Marcé RM. A quick, easy, cheap, effective, rugged and safe extraction method followed by liquid chromatography-(Orbitrap) high resolution mass spectrometry to determine benzotriazole, benzothiazole and benzenesulfonamide derivates in sewage sludge. J Chromatogr A. 2014;1339:34–41.
Naccarato A, Gionfriddo E, Sindona G, Tagarelli A. Simultaneous determination of benzothiazoles, benzotriazoles and benzosulfonamides by solid phase microextraction-gas chromatography-triple quadrupole mass spectrometry in environmental aqueous matrices and human urine. J Chromatogr A. 2014;1338:164–73.
Wang L, Asimakopoulos AG, Kannan K. Accumulation of 19 environmental phenolic and xenobiotic heterocyclic aromatic compounds in human adipose tissue. Environ Int. 2015;78:45–50.
Wan Y, Xue J, Kannan K. Benzothiazoles in indoor air from Albany, New York, USA, and its implications for inhalation exposure. J Hazard Mater. 2016;311:37–42.
Schlechtriem C, Fliedner A, Schäfers C. Determination of lipid content in fish samples from bioaccumulation studies: contributions to the revision of guideline OECD 305. Environ Sci Eur. 2012;24:13.
Smeraglia J, Baldrey SF, Watson D. Matrix effects and selectivity issues in LC-MS-MS. Chromatographia. 2002;55:95–9.
Herrero P, Borrull F, Pocurull E, Marcé RM. An overview of analytical methods and occurrence of benzotriazoles, benzothiazoles and benzenesulfonamides in the environment. Trends Anal Chem. 2014;62:46–55.
Caban M, Migowska N, Stepnowski P, Kwiatkowski M, Kumirska J. Matrix effects and recovery calculations in analyses of pharmaceuticals based on the determination of beta-blockers and beta-agonists in environmental samples. J Chromatogr A. 2012;1258:117–27.
Jover E, Matamoros V, Bayona JM. Characterization of benzothiazoles, benzotriazoles and benzosulfonamides in aqueous matrixes by solid-phase extraction followed by comprehensive two-dimensional gas chromatography coupled to time-of-flight mass spectrometry. J Chromatogr A. 2009;1216:4013–9.
ENCAT. Avaluació de l’estat nutricional de la població catalana 2002–2003. Enquesta Nutricional de Catalunya, Barcelona, Catalunya, Spain [in Catalan] 2003.
IPCS. International programme on chemical safety. Principles and methods for the risk assessment of chemicals in food. Environmental health criteria. 2009.
Trabalón L, Cano-Sancho G, Pocurull E, Nadal M, Domingo JL, Borrull F. Exposure of the population of Catalonia (Spain) to musk fragrances through seafood consumption: risk assessment. Environ Res. 2015;143:116–22.
European Commission. Introduction to risk assessment. 2003. http://ec.europa.eu/health/ph_projects/2003/action3/docs/2003_3_09_a23_en.pdf. Accessed 16 Oct 2016.
Hoff RB, Pizzolato TM, do CR PM, Díaz-Cruz MS, Barceló D. Determination of sulfonamide antibiotics and metabolites in liver, muscle and kidney samples by pressurized liquid extraction or ultrasound-assisted extraction followed by liquid chromatography–quadrupole linear ion trap-tandem mass spectrometry. Talanta. 2015;134:768–78.
Aznar-Alemany Ò, Trabalón L, Jacobs S, Barbosa VL, Tejedor MF, Granby K, Kwadijk C, Cunha SC, Ferrari F, Vandermeersch G, Sioen I, Verbeke W, Vilavert L, Domingo JL, Eljarrat E, Barceló D. Occurrence of halogenated flame retardants in commercial seafood species available in European markets. Food Chem Toxicol. 2016; doi:10.1016/j.fct.2016.12.034.
Ottonello G, Ferrari A, Magi E. Determination of polychlorinated biphenyls in fish: optimisation and validation of a method based on accelerated solvent extraction and gas chromatography-mass spectrometry. Food Chem. 2014;142:327–33.
Wang L, Asimakopoulos AG, Moon HB, Nakata H, Kannan K. Benzotriazole, benzothiazole, and benzophenone compounds in indoor dust from the United States and East Asian countries. Environ Sci Technol. 2013;47:4752–9.
Acknowledgments
The research leading to these results has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 311820.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Trabalón, L., Nadal, M., Borrull, F. et al. Determination of benzothiazoles in seafood species by subcritical water extraction followed by solid-phase microextraction-gas chromatography-tandem mass spectrometry: estimating the dietary intake. Anal Bioanal Chem 409, 5513–5522 (2017). https://doi.org/10.1007/s00216-017-0487-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0487-3