
Abstract
A rapid extraction procedure is presented for the determination of five endocrine-disrupting compounds, estrone, ethinylestradiol, bisphenol A, triclosan, and 2-ethylhexylsalicylate, in water samples. The analysis involves a two-step extraction procedure that combines dispersive liquid–liquid microextraction (DLLME) with dispersive micro-solid phase extraction (D-μ-SPE), using magnetic nanoparticles, followed by in situ derivatization in the injection port of a gas chromatograph coupled to triple quadrupole mass spectrometry. The use of uncoated or oleate-coated Fe3O4 nanoparticles as sorbent in the extraction process was evaluated and compared. The main parameters involved in the extraction process were optimized applying experimental designs. Uncoated Fe3O4 nanoparticles were selected in order to simplify and make more cost-effective the procedure. DLLME was carried out at pH 3, during 2 min, followed by the addition of the nanoparticles for D-μ-SPE employing 1 min in the extraction. Analysis of spiked water samples of different sources gave satisfactory recovery results for all the compounds with detection limits ranging from 7 to 180 ng l−1. Finally, the procedure was applied in tap, well, and river water.

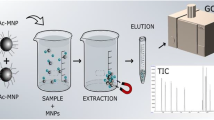
Diagram of the extraction method using magnetic nanoparticles (MNPs)
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Endocrine-disrupting compounds (EDCs) are naturally occurring or man-made substances that alter functions of the endocrine system and consequently cause adverse health effects in an intact organism, its progeny, or (sub)populations [1]. A wide range of substances, present in a high variety of everyday products, are thought to cause endocrine disruption at very low concentrations [2]. Additionally, a challenge to the field of endocrine disruption is that these substances are diverse and apparently do not share any structural similarity [3, 4]. The EDCs enter surface water from different sources such as direct discharge of industrial and domestic wastewater, effluents of wastewater treatment plants (WWTPs), and agricultural activities that cause nonpoint source pollution [4].
In this work, estrone (E1), ethinylestradiol (EE2), bisphenol A (BPA), triclosan (TCS), and 2-ethylhexylsalicylate (EHS) are analyzed. Steroid hormones are considered the most potent active EDCs present in the environment [5, 6]. E1 and EE2 are a natural and a synthetic estrogen, respectively. E1 is one of the main metabolites of estradiol that is excreted through human and animal urine into the environment. EE2 is currently included in the revised list of EU Priority substances in the field of water policy [7] due to its high estrogenic potency. BPA is an intermediate in the production of epoxy resins and polycarbonate plastics which is used as a model agent for endocrine disruption. It was described that perinatal exposure to environmentally relevant doses of this compound results in morphological and functional alterations [8]. This compound is included in the list of substances with endocrine disruption classifications with category 1 for wildlife and human health due to the evidence of endocrine-disrupting activity in at least one species using intact animals [9]. TCS is a broad-spectrum bactericide used in consumer products that could enter the environment by spreading sludge from WWTPs or through treated wastewater effluents. The last product included in our study is EHS, an ultraviolet (UV) filter. UV filters could enter the environment indirectly via WWTPs or directly via wash-off from the skin. EHS is one of the UV filters approved in cosmetic products in the European Union, which is classified as toxic to aquatic life with long-lasting effects [10].
Due to the low level of EDCs in environmental samples, the extraction, clean-up, and preconcentration of samples prior to their chromatographic analysis is necessary. Solid-phase extraction (SPE) is the extraction technique most commonly applied in the determination of steroid hormones in liquid samples. Nevertheless, miniaturized liquid–liquid extraction methods and other extraction methodologies, based on solid-phase microextraction (SPME), have also been used for the analyses of EDCs [11–14]. In recent years, some SPE methods using nanoparticles (NPs) of magnetite (Fe3O4) have been evaluated for the preconcentration of contaminants from liquid samples [15]. The use of magnetic nanostructures in the extraction of contaminants from liquid samples is a very interesting tool because the magnetic nanoparticles (MNPs) with the analytes are isolated by placing a magnet on the wall of the flask, discarding the solution, and the target compounds can be eluted from the sorbent with a low quantity of an adequate organic solvent to be analyzed. In the last years, a few magnetic solid-phase extraction (mSPE) methods have been described in the available literature for the analysis of different EDCs (phenols, UV filter, or some hormones) in water samples [16–23].
Recently, the application of MNPs in two-step microextraction techniques for liquid samples has been described. In these procedures, dispersive liquid–liquid microextraction (DLLME) combined with dispersive micro-solid phase extraction (D-μ-SPE), in which the retrieval of the extraction solvent is based on the adsorption by MNPs [24–28], was described as an alternative to the conventional DLLME. Thus, the organic phase containing the analytes is the target of the magnetic retrieval step rather than the analytes directly. After DLLME, MNPs retrieved 1-octanol from the aqueous solution via hydrophobic interaction. In the two-step extraction procedure, the need for a centrifugation or cooling step used in the conventional DLLME is overcome.
In general, most of the published studies concerning the presence of EDCs in water samples are focused on a limited number of compounds of a same group, being natural estrogens, alkylphenol, and BPA the most widely studied compounds. Nevertheless, the presence of a broad number of substances classified as EDCs has been recently investigated in surface and tap water [4, 29, 30]. In these studies, the multi-residue analysis was carried out by a sophisticated method using dual column liquid chromatography switching system coupled to tandem mass spectrometry.
Currently, quantification of EDCs in water samples has been mainly achieved by liquid chromatography–tandem mass spectrometry (LC–MS/MS) [4, 30–33]. Nevertheless, liquid chromatography–diode array detector (LC–DAD) [23], gas chromatography–mass spectrometry (GC–MS) [34, 35], or gas chromatography–tandem mass spectrometry (GC–MS/MS) [36–38] have also been reported. The choice of the analytical technique depends on the physico-chemical properties and concentrations of the compounds to be analyzed and, in many situations, on the equipment availability. Due to the high polarity, non-volatility, and thermal lability of many EDCs, a derivatization step before GC analysis is necessary to improve their volatility and chromatographic behavior. Silylation is one of the derivatization procedures widely used to improve GC behavior of polar compounds containing phenolic groups. Silylation of some EDCs using N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) and pyridine has been previously used by our group [39] and by other authors [35, 38], although other derivatization procedures had been reported in the available literature. The addition of pyridine, as derivatization reagent with BSTFA, has been reported to provide higher chromatographic responses, probably due to its catalytic and acid scavenging properties, and to prevent the conversion of EE2 to other products during the derivatization [35, 39]. Derivatization procedures are sometimes laborious and time consuming that often require the use of toxic reagents; thus, in situ derivatization in the GC injector is an attractive alternative because it avoids preparative steps, accelerates reaction rates, and reduces evaporative losses [37].
The main objective of this study was to develop a quick, cheap, and efficient method for the simultaneous analysis of EDCs in water samples. The method developed was based on DLLE combined with D-μ-SPE using MNPs, followed by in situ derivatization in the GC–MS/MS system. Finally, the validated method was used to monitor these contaminants in water samples from different sources.
Experimental
Reagents and standards
Ethyl acetate (EtAc), acetonitrile (ACN), n-hexane, and methanol (MeOH), residue analysis grade, were purchased from Scharlab (Spain). Fe (II, III) oxide MNPs (diameter 50–100 nm), sodium oleate (purity ≥95 %), sodium chloride (NaCl), and anhydrous sodium sulfate were acquired from Sigma-Aldrich (Steinheim, Germany). 1-Octanol was supplied by Merck (Germany). Chlorobenzene and carbon tetrachloride were obtained from Panreac (Spain). Chloroform was acquired from Fisher Chemical (United Kingdom). The derivatization agent BSTFA, purity 99 %, was from Supelco (Pennsylvania, USA). Pyridine, hydrochloric acid (37 %), and sodium hydroxide were purchased from Panreac (Spain). Ultrahigh-quality water was obtained from a Milli-Q water purification system (Millipore, Spain).
EHS, E1, and EE2 (all with purity >97 %) were obtained from Sigma–Aldrich (St Louis, MO, USA). TCS, purity >97 %, was purchased from Riedel-de Haën (Seelze, Germany), whereas BPA and the labeled compounds used as internal standards (13C12-TCS, 13C12-BPA, and 13C12-E1) were supplied by Cambridge Isotope Laboratories (Andover, MA, USA).
Working mixture solution containing all analytes (EHS, TCS, BPA, and E1 at 200 ng ml−1, and EE2 at 600 ng ml−1) and the internal standard solution containing 13C12-TCS, 13C12-BPA, and 13C12-E1 (at 200 ng ml−1) were prepared in MeOH. All standard solutions were stored at −20 °C prior to use.
Samples and MNPs
Samples were collected in 250-ml amber glass bottles. Tap water was collected from our laboratory. The other samples analyzed were well water and river water (Manzanares River) from Madrid. River samples were filtered through a paper filter (8 μm particle retention; Whatman) before the extraction procedure in order to remove the suspended solids.
Surface modification of the Fe3O4 MNPs with sodium oleate was carried out as described in our previous work [40]. Elemental analysis of the MNPs was performed with a LECO CHNS-932 microanalyzer at the Institute of General Organic Chemistry of the CSIC (Madrid, Spain).
Extraction procedure
The presented procedure is based on our previous work [28]. A mixture of 60 μl of 1-octanol and 4 ml of MeOH was quickly injected into a 100-ml conical flask containing 25 ml of sample adjusted to pH 3 with HCl 3 N. The sample was spiked with the internal standard solution, and it was closed with a glass cap. Then, the mixture was vigorously stirred using a vibromatic mixer at a vibration frequency of 782 oscillations min−1 for 2 min. Subsequently, 50 mg of MNPs was quickly added to the flask that was stirred for another 1 min. The MNPs were isolated by placing the magnet on the wall of the flask, and the supernatant was quickly discarded. Finally, the EDCs were extracted from the MNPs sonicating twice with 2 ml of EtAc each, during 1 min. The extract was evaporated using a Genevac EZ-2 evaporator (NET Interlab, S.A.L., Spain; www.net-interlab.es), and the residue was redissolved in 200 μl of EtAc. Then, a small spatula-tip full of anhydrous sodium sulfate was added to the organic extract to remove any water, and, finally, the extract was analyzed in the GC–MS/MS system.
For the recovery studies, samples were spiked with the working solution to reach concentrations of 1 or 0.5 ng ml−1 (for EE2, the concentration was tripled). In all experiments, the internal standard solution at 0.5 ng ml−1 was added to each water sample before extraction.
GC–MS/MS analysis
GC–MS/MS analysis was performed using the same chromatographic instrument, column, and analyte confirmation criteria than those reported in our previous works [28, 37, 39, 40]. In situ derivatization was done in the GC injector at 280 °C using two-layer sandwich injections drawing 1 μl of sample and 0.5 μl of the derivatization reagent (BSTFA/pyridine (1:1, v/v)) in pulsed splitless mode (pulsed pressure, 45 psi = 310 kPa for 1.5 min) with the splitless injector purge valve activated 1.5 min after sample injection. The column temperature was maintained at 60 °C for 1.5 min, then programmed at 25 °C min−1 to 300 °C and held for 1.5 min and set to analyze after a 6-min solvent delay. The total run time was 12.6 min.
The mass spectrometer was operated in electron impact (EI) ionization mode. Analysis was performed in the Multiple Reaction Monitoring (MRM) mode. A gain factor of 10 was set. Table 1 summarizes the retention times and the transitions with their optimal collision energy of each analyte.
The quantification of the studied compounds was based on their relative response factor to seven calibration standards in the range from 20 to 125 ng ml−1, except for EE2 that ranged from 60 to 375 ng ml−1. Each calibration level was spiked with labeled internal standards at 60 ng ml−1, taking into account a preconcentration factor of 125.
Statistical analysis
The data analyses were performed using the statistical package Statgraphics Plus, release 5.0 (Manugistics, Maryland, USA).
Results and discussion
Derivatization
The automated liquid sampler used in this work is capable of making multilayer (sandwich) injections of the silylation reagent and the sample, so the derivatization takes place in the GC inlet [37]. Multilayer injections applying two different organic solvents (ACN and EtAc) and a standard/reagent volume ratio (1:0.5, v/v) were assayed for the in situ derivatization. Both solvents provided similar chromatographic response for the majority of the compounds, and EtAc was selected in further analysis based on their lower cost (ACN is approximately 1.4 times more expensive than a similar-grade EtAc), higher volatility, and lower toxicity. The amount of derivatization reagents and the reaction time are greatly reduced when in situ derivatization is applied. Higher reaction efficiencies with less manipulation of the sample are achieved with this technique than with manual derivatization.
Characterization of the MNPs
Characterization of the Fe3O4 MNPs was reported in a previous paper [40]. The analysis by TEM showed that both uncoated and Ol-coated MNPs had cubic shape and variable sizes, sometimes higher than 100 nm, but not obvious morphological differences were observed. Although the grafted oleate layer was thin, its presence on the surface of the MNPs could be determined by DTA, taking into account the difference in the temperature of the exothermic transformation corresponding to the oxidative phase that were higher when the MNPs were coated, due to their higher thermal stability [40].
Additionally, the elemental analysis was employed to confirm the presence of oleate in the modified MNPs. The analyses showed a mass percent of carbon of 0 and 0.7 % in the unmodified and the modified MNPs, respectively. The presence of this element in the modified magnetite particles is attributed to the coating of oleate on the Fe3O4 MNPs surface. Although the low percentage obtained confirmed that the grafted oleate layer was very thin, probably due to the high size of the MNPs, it has been described that the oleate adsorption decreases with the increasing size of Fe3O4 MNPs [41].
Extraction procedure
The combination of DLLME with D-μ-SPE using uncoated and Ol-coated MNPs as sorbents was evaluated. The use of MNPs overcomes the need for a centrifugation or cooling step used in conventional DLLME procedures. In preliminary studies, 1-octanol was selected as the extractant because it is one of the most used organic solvents in liquid phase microextraction, it is less toxic than conventional halogenated hydrocarbons, and is immiscible with water and free from loss during agitation (due to its low vapor pressure and viscosity) [42]. Although 1-octanol is not generally applied in classical DLLME, it has been reported as a good solvent to be extracted and retrieved from an aqueous sample by D-μ-SPE [25–27]. The DLLME using 1-octanol could be carried out with or without a disperser solvent. Thus, in a first assay, the DLLME was done with 50 μl of 1-octanol, quickly injected and vigorously stirred for 2 min, and the results were compared with those obtained when the quick injection was done with 4 ml of MeOH, as disperser solvent, containing 50 μl of 1-octanol. In all the assays, the D-μ-SPE step was carried out with 50 mg of MNPs (uncoated and Ol-coated) stirring at high speed for 2 min. The recoveries obtained were in general better when MeOH was used as disperser solvent. When uncoated MNPs were used, recoveries >40 % for EHS and E1 and >82 % for TCS, BPA, and EE2 were obtained, and when Ol-coated MNPs were applied, recoveries >51 % for EHS and E1 and >90 % for the rest were achieved (see Fig. 1). Although the results obtained in this preliminary assay seem to show certain improvement in the recoveries with Ol-coated MNPs, these differences were not significant to select the Ol-coated MNPs. Therefore, optimization of the main parameters of the extraction process was carried out using both uncoated and coated MNPs.

Recoveries obtained with and without dispersive solvent in the DLLME step using A uncoated Fe3O4 NPs and B Ol-coated Fe3O4 NPs in the D-μ-SPE. Number of replicates n = 3
The selection of a right extraction solvent is an essential parameter in DLLME. For this reason, extraction solvents with different densities, carbon tetrachloride, chlorobenzene, chloroform, and 1-octanol were assayed using MeOH (4 ml) as disperser solvent and uncoated MNPs (50 mg) to retrieve the extraction solvent. The results showed that the best extraction capacities were obtained with 1-octanol, followed by chlorobenzene, whereas with chloroform and carbon tetrachloride, they were very low (data not shown). According to this, the extraction capacity of 1-octanol and chlorobenzene using Ol-coated MNPs was evaluated, and similar or better results were obtained with 1-octanol.
Once the optimal DLLME solvent was selected, the effect of the amount of MNPs and extraction time were evaluated. The response surface methodology has already proven to be a reliable statistical tool in the investigation of chemical treatment processes to optimize parameters with a minimal number of experiments. Therefore, a statistical analysis was done to improve the efficiency of the procedure, and the effect of the amount of MNPs and different volumes of 1-octanol were simultaneously studied using a multilevel experimental factorial design to obtain an optimal response. Thus, these parameters were optimized for uncoated and Ol-coated MNPs, applying a 32 experimental design at three levels and five factors that allows estimating the influence of two factors simultaneously on the recoveries. The 1-octanol volumes were 25, 50, and 75 μl, and the amounts of uncoated or coated NPs were 30, 50, and 75 mg. In general, 1-octanol volume significantly affected the extraction efficiency, whereas the amount of MNPs did not show significant differences. Nevertheless, the responses depended on the analyte. EHS showed a high negative significant dependence with the volume of extractant, whereas this high dependence was positive for E1 and EE2 and no dependence was observed for TCS. The results obtained for BPA depended on the MNP used; therefore, positive significant influence or no statistically significant influence was obtained with uncoated and Ol-coated MNPs, respectively. According to this, the optimal response was very different for EHS than for the rest of the compounds (see Table 2a). Considering these results, as compromise for the whole group of EDCs evaluated, a 1-octanol volume of 60 μl and 50 mg of MNPs were selected for further assays. The first-order interactions obtained in this study for two representative compounds (EHS and E1) are summarized in the Pareto charts, and the response surfaces shown in Fig. 2a.

Pareto charts and response surfaces for understanding the recovery (%) of EHS and E1 as function on the effect of volume of octanol and MNPs quantity (A) and as function of the effect of pH and extraction time (B), using uncoated Fe3O4 MNPs
Two other important factors were the pH for the DLLME and the extraction time for D-μ-SPE. The effects of these two parameters were evaluated using a multilevel experimental factorial design, a 32 experimental design at three levels and six factors. The pH values were 2.5, 6.5, and 10, and the extraction times were 2, 5.5, and 10 min. The results obtained using uncoated and Ol-coated MNPs showed that pH significantly affects the efficiency of the extraction and the optimum was pH 3. This result is in agreement with those reported by other authors that extract EDCs from water by mSPE or DLLME at pH from 2 to 4 [17, 43–46]. In general, extraction time was found to have no effect on EDC recoveries (see Table 2b). As example, Fig. 2b shows the Pareto charts and the response surfaces obtained for EHS and E1. In general, an extraction time of 1 min was appropriate to obtain the optimum recovery. Taking into consideration the standard response, further analyses were carried out at pH 3 and with an extraction time of only 1 min.
During the optimization, uncoated and Ol-coated MNPs were assayed. The results showed again that the efficiency of both MNPs retrieving the extractive solvent was very similar. Given that Ol-coated MNPs were obtained from uncoated Fe3O4 MNPs, it should be noted that the surface modification involves spending time, reagents, and handling. Therefore, in order to simplify the process and to make it much more cost-effective, further analyses and validation of the method were carried out using only uncoated Fe3O4 MNPs.
Additionally, the effect of ionic strength was investigated by adding NaCl in the range of 0 to 15 %. Results showed that recoveries did not increase in the presence of NaCl, and a significant decrease in the recovery of EHS was obtained after salt addition (data not shown). Therefore, salt was not added in the following assays. Finally, desorption of the analytes from the MNPs was performed by sonication. EtAc and ACN were examined for their suitability as desorption solvents in the post-DLLME/μ-SPE step. Results showed that EtAc gave the highest overall extraction efficiency for EHS and TCS, and similar than ACN for BPA, E1, and EE2. However, as described above, EtAc provided similar chromatographic response than ACN in the in situ derivatization in the GC injector. Therefore, EtAc was selected as the desorption solvent for the subsequent studies.
Based on the results shown above, the summary of the final conditions for the extraction procedure were as follows: 25 ml of sample, at pH 3, extraction during 2 min with 1-octanol (60 μl) as the extraction solvent with MeOH as disperser solvent, followed by the addition of 50 mg of MNPs, and 1 min of extraction time for D-μ-SPE, and, finally, desorption from the MNPs using EtAc.
Method validation
After optimization, the method was evaluated in terms of linearity, precision, accuracy, and detection limits before it was used to determine EDCs in water samples.
The linearity of the method was evaluated injecting seven standard solutions prepared at 20, 30, 40, 50, 65, 80, and 125 ng ml−1 levels for all the studied compounds except for EE2 that were prepared at triple concentration due to their low chromatographic response. The set of standards contained the internal standards at 60 ng ml−1. A good linearity was obtained, with correlation coefficients equal or higher than 0.98 for all the compounds studied.
The quality assurance and quality control criteria used for this method included analyses of laboratory blanks (solvent blank) and laboratory control samples (LCS) with each set of samples. One laboratory blank was run with each set of samples to check potential contamination from the preparative steps and to demonstrate laboratory background levels. LCS were used in the recovery assays. Quantification was carried out with internal standard calibration to avoid the dependence of the results with the sample matrix, improving the reproducibility and accuracy of the analyses. The accuracy of the method was evaluated performing the recovery of target analytes from water samples of Milli-Q water, spiked at two concentrations, 1 and 0.5 ng ml−1, using isotopically labeled compounds as surrogate standard 0.5 ng ml−1. Table 3 shows the satisfactory recovery results obtained (>78 %) for all the compounds at the levels assayed.
The repeatability of the chromatographic determination was determined by injecting ten times a fortified blank of water sample within a given day. The precision (expressed as relative standard deviations, RSDs) were <10 %. Within-laboratory reproducibility of the chromatographic determination was evaluated during different days and was found to be lower than 13 % for all the compounds, expressed as RSD. The limits of detection (LODs) and limits of quantification (LOQs) of the developed method were calculated by GC–MS/MS after the analysis of eight replicates of water spiked at 0.24 ng ml−1. These values were obtained following the t99sLLMV approach developed by EPA [47]. As shown in Table 3, LODs for pure water ranged from 7 to 138 ng l−1.
Real samples
In order to assess the suitability of the developed DLLME/D-μ-SPE method for the analysis of real samples, three different surface water samples of different matrix composition (tap, river, and well) were studied. Thus, once the method was validated for pure water, it was validated for these environmental waters. Reagent and sample blanks were prepared together with the water samples analyzed to check for contamination and EDC presence, respectively. Table 4 shows the recovery results obtained that ranged between 67 and 109 %, with relative standard deviations <11 %. The efficiency obtained by DLLME/D-μ-SPE was in the range reported in other studies (Table 5). LOD values of tap, well, and river waters were slightly higher than those obtained for pure water, ranging from 22 to 180 ng l−1. Although the values obtained for TCS or EHS are somewhat higher than those obtained by mSPE [44] or SPE combined with DLLME [17, 44], respectively, in general, the LODs obtained by DLLME/D-μ-SPE are similar to or lower than those reported by other authors for the determination of EDCs from water samples (see Table 5). Thus, LODs of about 1000 ng ml−1 were reported for the analysis of the estrogenic compounds by DLLME integrated with the solidification of a floating organic drop [48]; LODs of 890 ng ml−1 for BPA extracted from water samples by mSPE was reported by Meng et al. [49], and the limit obtained for EHS by Zhang and Lee [46] with ionic liquid-based ultrasound-assisted DLLME was 5000 ng l−1. LODs of 8 ng l-1 for E1 extracted from water samples were obtained by mSPE using 200 mg of palmitate coated NPs (PA-Fe3O4 NPs) [50].
The method developed was applied in different water samples: tap, well, and river water. None of the five EDCs were detected in unspiked tap water, but BPA was found in well water and river water at 189 and 268 ng l−1, respectively. High concentrations of BPA in river water (7940 ng l−1) were found by Meng et al. [49]; however, Cunha et al. [45] only detected BPA in effluent waters in 4 out of 15 WWTPs evaluated with concentrations between 61 and 874 ng l−1. The presence of EHS was described by Benedé et al. [43] in three seawater samples at levels from 440 to 880 ng l−1 and by Roman et al. [17] in tap and river water at 160 and 146 ng l−1, respectively. Nevertheless, this UV filter was not detected or was only some time detected in river water or wastewaters [44, 45, 51]. Zheng et al. [52] did not find TCS in tap water, but concentrations between 26 and 38 ng l−1 were detected in river or lake waters. Finally, estrogens were not found using any of the analytical methods reported in Table 5.
Conclusions
In the present study, a two-step microextraction technique based on DLLME and D-μ-SPE using MNPs was developed for the extraction of five EDCs from surface water. In situ derivatization carried out in the GC system was used as an attractive alternative to manual procedures because it simplifies the sample preparation and avoids the manipulation of hazardous reagents. The effect of several parameters of the DLLME/D-μ-SPE method was analyzed using uncoated and Ol-coated MNPs, and it was observed that the efficiencies of both MNPs retrieving the extractive solvent were very similar. Thus, in order to simplify the process and to make it much more cost-effective, the extraction was performed using uncoated MNPs. The method was applied to different water samples, and the recovery results obtained were satisfactory for all the compounds. The analyses confirmed the presence BPA in well and river waters. The DLLME/D-μ-SPE method developed is an easy and quick method that only required about 5 min for the extraction procedure and does not need any special laboratory equipment to be carried out.
References
WHO/IPCS (World Health Organization/International Petroleum Chemical Society). In: Damstra T, Barlow S, Bergman A, Kavlock R, Van Der Kraak G, editors. Global assessment of the state-of-the-science of endocrine disruptors. WHO/IPCS/EDC/02.2. Geneva: World Health Organization; 2002. http://ehp.niehs.nih.gov/who/.
Snow DD, Damon-Powell T, Onanong S, Cassada DA. Sensitive and simplified analysis of natural and synthetic steroids in water and solids using on-line solid-phase extraction and microwave-assisted solvent extraction coupled to liquid chromatography tandem mass spectrometry atmospheric pressure photoionization. Anal Bioanal Chem. 2013;405:1759–71.
Diamanti-Kandarakis E, Bourguignon J-P, Giudice LC, Hauser R, Prins GS, Soto AM, et al. Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocr Rev. 2009;30:293–342.
Gorga M, Insa S, Petrovic M, Barceló D. Occurrence and spatial distribution of EDCs and related compounds in waters and sediments of Iberian rivers. Sci Total Environ. 2015;503:69–86.
Ingerslev F, Halling-Sorensen B. Evaluation of analytical methods for detection of estrogens in the environment. Working Report No 44. Copenhagen: Danish Environmental Protection Agency; 2003.
Sosa-Ferrera Z, Mahugo-Santana C, Juan Santana-Rodriguez J. Analytical methodologies for the determination of endocrine disrupting compounds in biological and environmental samples. Biomed Res Int. 2013;Article ID 674838, 23 pages, doi:10.1155/2013/674838.
European Commission Endocrine Disruptors priority lists Annex 13. List of 146 substances with endocrine disruption classifications prepared in the Expert meeting http://ec.europa.eu/environment/archives/docum/pdf/bkh_annex_13.pdf
Maffini MV, Rubin BS, Sonnenschein C, Soto AM. Endocrine disruptors and reproductive health: the case of bisphenol-A. Mol Cell Endocrinol. 2006;254:179–86.
European Commission COM/2011/0876 final—2011/0429 (COD) Proposal for a DIRECTIVE OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy.
Sobek A, Bejgarn S, Ruden C, Molander L, Breitholtz M. In the shadow of the Cosmetic Directive—inconsistencies in EU environmental hazard classification requirements for UV-filters. Sci Total Environ. 2013;461:706–11.
Tomsikova H, Aufartova J, Solich P, Sosa-Ferrera Z, Santana-Rodriguez J, Novakova L. High-sensitivity analysis of female-steroid hormones in environmental samples. TrAC-Trends Anal Chem. 2012;34:35–58.
Pacakova V, Loukotkova L, Bosakova Z, Stulik K. Analysis for estrogens as environmental pollutants—a review. J Sep Sci. 2009;32:867–82.
Li Q, Wang X, Yuan D. Preparation of solid-phase microextraction fiber coated with single-walled carbon nanotubes by electrophoretic deposition and its application in extracting phenols from aqueous samples. J Chromatogr A. 2009;1216:1305–11.
Canosa P, Rodriguez I, Rubi E, Cela R. Optimization of solid-phase microextraction conditions for the determination of triclosan and possible related compounds in water samples. J Chromatogr A. 2005;1072:107–15.
Xie L, Jiang R, Zhu F, Liu H, Ouyang G. Application of functionalized magnetic nanoparticles in sample preparation. Anal Bioanal Chem. 2014;406:377–99.
Alcudia-Leon MC, Lucena R, Cardenas S, Valcarcel M. Magnetically confined hydrophobic nanoparticles for the microextraction of endocrine-disrupting phenols from environmental waters. Anal Bioanal Chem. 2013;405:2729–34.
Roman IP, Chisvert A, Canals A. Dispersive solid-phase extraction based on oleic acid-coated magnetic nanoparticles followed by gas chromatography–mass spectrometry for UV-filter determination in water samples. J Chromatogr A. 2011;1218:2467–75.
Zhao XL, Shi YL, Ca YQ, Mou SF. Cetyltrimethylammonium bromide-coated magnetic nanoparticles for the preconcentration of phenolic compounds from environmental water samples. Environ Sci Technol. 2008;42:1201–6.
Jiang X, Cheng J, Zhou H, Li F, Wu W, Ding K. Polyaniline-coated chitosan-functionalized magnetic nanoparticles: preparation for the extraction and analysis of endocrine-disrupting phenols in environmental water and juice samples. Talanta. 2015;141:239–46.
Liu Y, Jia L. Analysis of estrogens in water by magnetic octadecylsilane particles extraction and sweeping micellar electrokinetic chromatography. Microchem J. 2008;89:72–6.
Ye L, Wang Q, Xu J, Shi ZG, Xu L. Restricted-access nanoparticles for magnetic solid-phase extraction of steroid hormones from environmental and biological samples. J Chromatogr A. 2012;1244:46–54.
Liu L, Feng T, Wang C, Wu Q, Wang Z. Magnetic three-dimensional graphene nanoparticles for the preconcentration of endocrine-disrupting phenols. Microchim Acta. 2014;181:1249–55.
Li F, Cai C, Cheng J, Zhou H, Ding K, Zhang L. Extraction of endocrine disrupting phenols with iron-ferric oxide core-shell nanowires on graphene oxide nanosheets, followed by their determination by HPLC. Microchim Acta. 2015;182:2503–11.
Tay KS, Abd Rahman N, Bin Abas MR. Magnetic nanoparticle assisted dispersive liquid-liquid microextraction for the determination of 4-n-nonylphenol in water. Anal Methods. 2013;5:2933–8.
Shi ZG, Lee HK. Dispersive liquid-liquid microextraction coupled with dispersive μ-solid-phase extraction for the fast determination of polycyclic aromatic hydrocarbons in environmental water samples. Anal Chem. 2010;82:1540–5.
Mukdasai S, Thomas C, Srijaranai S. Enhancement of sensitivity for the spectrophotometric determination of carbaryl using dispersive liquid microextraction combined with dispersive μ-solid phase extraction. Anal Methods. 2013;5:789–96.
Wang N, Shen R, Yan Z, Feng H, Cai Q, Yao S. Magnetic retrieval of an extractant: fast ultrasound assisted emulsification liquid-liquid microextraction for the determination of polycyclic aromatic hydrocarbons in environmental water samples. Anal Methods. 2013;5:3999–4004.
Pérez RA, Albero B, Tadeo JL, Sánchez-Brunete C. Oleate functionalized magnetic nanoparticles as sorbent for the analysis of polychlorinated biphenyls in juices. Microchim Acta. 2016;183:157–65.
Esteban S, Gorga M, Gonzalez-Alonso S, Petrovic M, Barceló D, Valcarcel Y. Monitoring endocrine disrupting compounds and estrogenic activity in tap water from Central Spain. Environ Sci Pollut R. 2014;21:9297–310.
Gorga M, Petrovic M, Barceló D. Multi-residue analytical method for the determination of endocrine disruptors and related compounds in river and waste water using dual column liquid chromatography switching system coupled to mass spectrometry. J Chromatogr A. 2013;1295:57–66.
Esteban S, Gorga M, Petrovic M, Gonzalez-Alonso S, Barceló D, Valcarcel Y. Analysis and occurrence of endocrine-disrupting compounds and estrogenic activity in the surface waters of Central Spain. Sci Total Environ. 2014;466:939–51.
Carvalho ARM, Cardoso VV, Rodrigues A, Ferreira E, Benoliel MJ, Duarte EA. Quality assessment of sulfurous thermal waters in the city of Poços de Caldas, Minas Gerais, Brazil. Environ Monit Assess. 2015;187(139):1–8.
Martin J, Santos JL, Aparicio I, Alonso E. Determination of hormones, a plasticizer, preservatives, perfluoroalkylated compounds, and a flame retardant in water samples by ultrasound-assisted dispersive liquid-liquid microextraction based on the solidification of a floating organic drop. Talanta. 2015;143:335–43.
Azzouz A, Ballesteros E. Trace analysis of endocrine disrupting compounds in environmental water samples by use of solid-phase extraction and gas chromatography with mass spectrometry detection. J Chromatogr A. 2014;1360:248–57.
Zhang ZL, Hibberd A, Zhou JL. Optimisation of derivatisation for the analysis of estrogenic compounds in water by solid-phase extraction gas chromatography-mass spectrometry. Anal Chim Acta. 2006;577:52–61.
Mudiam MKR, Jain R, Singh R. Application of ultrasound-assisted dispersive liquid-liquid microextraction and automated in-port silylation for the simultaneous determination of phenolic endocrine disruptor chemicals in water samples by gas chromatography-triple quadrupole mass spectrometry. Anal Methods. 2014;6:1802–10.
Albero B, Sánchez-Brunete C, Miguel E, Perez RA, Tadeo JL. Determination of selected organic contaminants in soil by pressurized liquid extraction and gas chromatography tandem mass spectrometry with in situ derivatization. J Chromatogr A. 2012;1248:9–17.
Hibberd A, Maskaoui K, Zhang Z, Zhou JL. An improved method for the simultaneous analysis of phenolic and steroidal estrogens in water and sediment. Talanta. 2009;77:1315–21.
Albero B, Sánchez-Brunete C, Miguel E, Pérez RA, Tadeo JL. Analysis of natural-occurring and synthetic sexual hormones in sludge-amended soils by matrix solid-phase dispersion and isotope dilution gas chromatography-tandem mass spectrometry. J Chromatogr A. 2013;1283:39–45.
Pérez RA, Albero B, Tadeo JL, Molero E, Sánchez-Brunete C. Application of magnetic iron oxide nanoparticles for the analysis of PCBs in water and soil leachates by gas chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2015;407:1913–24.
Zhang L, He R, Gu H-C. Oleic acid coating on the monodisperse magnetite nanoparticles. Appl Surf Sci. 2006;253:2611–7.
Li Y, Yang X, Zhang J, Li M, Zhao X, Yuan K, et al. Ultrasound-assisted emulsification magnetic microextraction: a fast and green method for the determination of triazole fungicides in fruit juice. Anal Methods. 2014;6:8328–36.
Benedé JL, Chisvert A, Salvador A, Sánchez-Quiles D, Tovar-Sánchez A. Determination of UV filters in both soluble and particulate fractions of seawaters by dispersive liquid–liquid microextraction followed by gas chromatography–mass spectrometry. Anal Chim Acta. 2014;812:50–8.
Celano R, Piccinelli AL, Campone L, Rastrelli L. Ultra-preconcentration and determination of selected pharmaceutical and personal care products in different water matrices by solid-phase extraction combined with dispersive liquid–liquid microextraction prior to ultra high pressure liquid chromatography tandem mass spectrometry analysis. J Chromatogr A. 2014;1355:26–35.
Cunha SC, Pena A, Fernandes JO. Dispersive liquid-liquid microextraction followed by microwave-assisted silylation and gas chromatography-mass spectrometry analysis for simultaneous trace quantification of bisphenol A and 13 ultraviolet filters in wastewaters. J Chromatogr A. 2015;1414:10–21.
Zhang YF, Lee HK. Ionic liquid-based ultrasound-assisted dispersive liquid–liquid microextraction followed high-performance liquid chromatography for the determination of ultraviolet filters in environmental water samples. Anal Chim Acta. 2012;750:120–6.
Bernal E (2014) Limit of detection and limit of quantification determination in gas chromatography. In: Guo X, editor. Advances in gas chromatography. Intech, 2014, doi: 10.5772/57341
Chang C-C, Huang S-D. Determination of the steroid hormone levels in water samples by dispersive liquid-liquid microextraction with solidification of a floating organic drop followed by high-performance liquid chromatography. Anal Chim Acta. 2010;662:39–43.
Meng J, Shi C, Wei B, Yu W, Deng C, Zhang X. Preparation of Fe3O4@C@PANI magnetic microspheres for the extraction and analysis of phenolic compounds in water samples by gas chromatography-mass spectrometry. J Chromatogr A. 2011;1218:2841–7.
Pérez RA, Albero B, Tadeo JL, Molero E, Sánchez-Brunete C. Analysis of steroid hormones in water using palmitate-coated magnetite nanoparticles solid-phase extraction and gas chromatography-tandem mass spectrometry. Chromatographia. 2014;77:837–43.
Wu JW, Chen HC, Ding WH. Ultrasound-assisted dispersive liquid–liquid microextraction plus simultaneous silylation for rapid determination of salicylate and benzophenone-type ultraviolet filters in aqueous samples. J Chromatogr A. 2013;1302:20–7.
Zheng C, Zhao J, Bao P, Gao J, He J. Dispersive liquid-liquid microextraction based on solidification of floating organic droplet followed by high-performance liquid chromatography with ultraviolet detection and liquid chromatography–tandem mass spectrometry for the determination of triclosan and 2,4-dichlorophenol in water samples. J Chromatogr A. 2011;1218:3830–6.
Acknowledgments
This study was financed by the Ministry of Science and Innovation–National Institute for Agricultural and Food Research and Technology, INIA, Project number RTA2014-00012-C03-01. The authors wish to express their gratefulness to Dr. M.P. Morales, researcher of the Department of Biomaterials and Bioinspired Materials (ICMM; CSIC), for her help and advice in the characterization of the MNPs and to the microanalysis laboratory of the Institute of General Organic Chemistry (CSIC) for the elemental analysis of the MNPs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Pérez, R.A., Albero, B., Tadeo, J.L. et al. Determination of endocrine-disrupting compounds in water samples by magnetic nanoparticle-assisted dispersive liquid–liquid microextraction combined with gas chromatography–tandem mass spectrometry. Anal Bioanal Chem 408, 8013–8023 (2016). https://doi.org/10.1007/s00216-016-9899-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9899-8