
Abstract
In this study, a system of magnetic beads (MBs) coupled with catalytic fluorescent immunoassay for rapid and sensitive determination of HIV-1 capsid antigen p24 was developed. p24 was captured by antibody immobilized MBs, and the detection antibody was linked to horseradish peroxidase (HRP) through biotin–streptavidin recognition, catalyzing the oxidation of o-phenylenediamine (OPD) and hydrogen peroxide to produce a fluorescent product. This is the first reported utilization of the fluorescence of OPD oxidation product catalyzed by HRP for immunoassay. Optimization of conditions afforded a low detection limit of 0.5 pg/mL (3σ) for p24 with a linear range of 1.4–90.0 pg/mL. The assay exhibited good reproducibility with a relative standard deviation (RSD) of 4.4 %, 4.7 %, and 5.0 % for detecting 1.4 pg/mL, 22.5 pg/mL, and 45.0 pg/mL p24, respectively. The assay can be completed in less than 90 min. Moreover, the proposed method was successfully applied to detect p24 in spiked serum. This method overcomes the interference of MBs to the fluorescence signal and demonstrated higher sensitivity for detection of p24 than conventional ELISA kits. The system could be applied for detecting other antigens with high sensitivity, rapidity, specificity, and simple operation.

A rapid and sensitive biosensing method coupling immunomagnetic separation and catalytic fluorescence for determination of HIV-1 p24 has been developed
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human immunodeficiency virus (HIV), which causes acquired immunodeficiency syndrome (AIDS), is a member of the lentivirus family. HIV destroys the cells of the immune system and impairs their function [1–3]. HIV is divided into HIV-1 and HIV-2, and HIV-1 is by far the most common cause of AIDS [4, 5]. Rapid, accurate, and early diagnosis of HIV infection is a key issue to control AIDS.
Diagnosis of HIV-1 infection is mainly focused on the detection of HIV-1 RNA, capsid antigens (such as p24), and anti-HIV antibodies [6–9]. PCR-based nucleic acid testing (NAT) is sensitive but expensive and complex, thus limiting its application [10–12]. HIV-1 p24 antigen in serum or plasma is one of the surrogate biomarkers for early diagnosis of HIV-1 infection, because it appears at an earlier stage of HIV infection than antibodies and remains at a high level during the early, acute phase of the infection [13–15]. Up to now, a number of detection systems have been investigated for the determination of p24, such as enzyme-linked immunosorbent assay (ELISA) [16], chemiluminescent immunoassay (CLIA) [17], electrochemical immunoassay [18–20], and nanoparticle-based assays [21–23]. However, most of these methods either are expensive, time-consuming, have low sensitivity, require rare metals, afford inherently inaccurate quantification of target molecules, or rely on complicated instruments and professional staff. For example, a plasmonic ELISA using enzyme-catalyzed growth of gold nanoparticles could detect a low concentration of 1 × 10−6 pg/mL p24, but needed a long analysis time [21], which would not keep pace with the requirements of future point-of-care testing. An inductively coupled plasma mass spectrometry based on ELISA and AuNPs coloration was able to detect a minimum of 1.49 pg/mL p24 [23], but relied on an expensive mass spectrometer. Hence, flexible, time-saving, sensitive, and easy-to-use approaches are still required for the detection of p24.
Magnetic beads (MBs)-based immunoassay has been widely accepted for separating biomolecules from complex matrix, because it can offer high target-capture efficiency, fast target-binding kinetics, and simple processing procedures [24–27]. These characteristics lead to increased sensitivity and short assay times for detecting a variety of targets in crude samples [28–30]. Fluorescence-based assay is sensitive, fast, and stable [31–33], but there are few fluorescent probes developed for antigen detection [34]. What is more, the autofluorescence of magnetic beads would interfere with fluorescence signals and lower the sensitivity of immunoassays, so an additional “disassociate and elute” tactic is normally adopted before collecting fluorescence signal [35–37], which would complicate the detection procedure and prolong the analysis time.
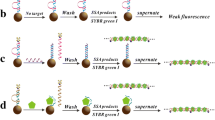
With the intention to address the problems mentioned above, we developed an MBs combining with catalytic fluorescent immunoassay platform for the rapid and sensitive detection of p24 based on horseradish peroxidase (HRP), which is a common labeling enzyme in conventional ELISA, owing to its high catalytic activity and excellent stability [38]. Moreover, it has been found that in the presence of HRP, the nonfluorescent substrate o-phenylenediamine (OPD) could be oxidized into a stable fluorescent product, 2,3-diamiophenazine (OPDox) [34, 39]. This was the first reported utilization of the fluorescence of OPD oxidation product catalyzed by HRP for immunoassay. A sandwich-type immunoassay format (Fig. 1) was adopted to detect p24 using streptavidin-labeled HRP linked with biotinylated polyclonal antibody to p24 as an enzyme tracer and OPD/H2O2 as the substrates. After the catalytic reaction, immunomagnetic beads (IMBs) were removed from the solution by magnetic separation, which avoided the interference from the magnetic beads on the fluorescence of the system. The proposed method was rapid, sensitive, and provides promising potential for antigen detection.

Schematic representation of the proposed magnetic beads coupled with catalytic fluorescent assay for the detection of HIV-1 p24 antigen (not to scale)
Experimental
Materials and reagents
Isothiocyanate-terminated magnetic beads (ITC-MBs) (3 μm in diameter) and magnetic separation racks were purchased from GoldMag Biotech Co., Ltd. (Xi’an, China). HIV-1 recombinant p24 antigen, biotin-rabbit anti-p24 polyclonal antibody (biotin–PcAb) (1 mg/mL), and mouse anti-p24 monoclonal antibody (McAb) (1 mg/mL) were gifts from Prof. Yan Bing, Wuhan Institute of Virology, Chinese Academy of Science. Human alpha-fetoprotein (AFP), human carcinoembryonic antigen (CEA), and human serum albumin (HSA) were purchased from Kitgen Biotech Co., Ltd (Hangzhou, China). HRP and Tween-20 were obtained from Sigma-Aldrich Chemicals Co. (St. Louis, MO, USA). Streptavidin-labeled HRP (1 mg/mL) was supplied by Beijing Cowin bioscience Co., Ltd. (Beijing, China). Human serum were obtained from Fankel Biological Technology Co., Ltd. (Shanghai, China). Skim milk powder was purchased from Oxoid Co., Ltd. (England), and bovine serum albumin (BSA) was supplied by German Roche Co. Ltd. (Germany). o-Phenylenediamine (OPD) was bought from Aladdin reagent Co., Ltd. (Shanghai, China). H2O2 was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Phosphate-buffered saline (PBS, pH 7.4) was prepared from 8 g sodium chloride, 0.24 g potassium chloride, 1.15 g disodium hydrogen phosphate, and 0.2 g potassium dihydrogen phosphate dissolving in 1000 mL distilled water. PBST (pH 7.4) was prepared by mixing 500 μL of Tween-20 with 1000 mL 0.01 mol/L PBS (pH 7.4). All other reagents were of analytical grade. Doubly distilled water (18.2 Ω) was used for all experiments.
Apparatus
A Synergy™ H1 multi-mode microplate reader (Biotek, USA) was utilized for scanning fluorescence spectroscopy and fluorescence measurements. A UV-3600 spectrophotometer (Shimadzu, Japan) was applied to record UV–visible spectra. A shaking incubator ZWY-200D (Labwit scientific, Co., Ltd., Shanghai, China) was used for the incubation. An Oscillator Vortex Genie 2 (USA) was used to disperse magnetic beads.
Preparation of immunomagnetic beads
ITC-MBs are ready-to-use superparamagnetic beads, the ITC groups of which are used to covalently bond to amino groups of proteins. In accordance with our previous report [40], IMBs were prepared by covalently binding McAb onto the superparamagnetic ITC-MBs. Briefly, 200 μL McAb (1 mg mL−1) was shaken (180 rpm) with 1 mg ITC-MBs at 37 °C for 30 min. After magnetic separation, the supernatant was discarded and the IMBs were washed twice with 200 μL of PBST to remove the unbound antibody. The prepared IMBs were shaken (180 rpm) in PBS containing 5 % bovine serum albumin (BSA) and 15 % skim milk at 37 °C for 2 h to block the sites which were not covered with McAb. The IMBs were washed twice to remove the residual blocking buffer. After separation on the magnetic separation racks, IMBs were resuspended in PBS containing 0.1 % NaN3 and 0.1 % BSA and stored at 4 °C until use.
Detection procedure
To detect p24, IMBs were added into a mixture of p24 (75 μL) and biotin–PcAb (25 μL) first. Then the reaction mixture was shaken (180 rpm) at 37 °C for 30 min. Afterwards the supernatant was removed after magnetic separation, and the immuno-complex was washed five times with PBST. Following that, 100 μL streptavidin-labeled HRP was added to resuspend the MBs. The mixture was incubated with rotation at 37 °C for 30 min. Finally, the immuno-complex (MBs/McAb/p24/biotin–PcAb/streptavidin-labeled HRP) was washed five times with PBST. Following the immune reaction mentioned above, the substrates OPD and H2O2 were mixed with the immuno-complex (MBs/McAb/p24/biotin–PcAb/streptavidin-labeled HRP). After incubation at room temperature for 10 min in darkness, the MBs were separated from the solution by a magnet. The fluorescence of the supernatant was measured using the microplate reader with an excitation wavelength of 398 nm and emission wavelength of 580 nm for quantitative measurement of p24.
Results and discussion
Characterization of HRP–OPD–H2O2 fluorescence system
In the presence of HRP, the nonfluorescent substrate OPD was efficiently oxidized by hydrogen peroxide into the fluorescent product OPDox, with a stable fluorescent emission at 580 nm (Fig. 2a). The kinetic characteristics of this catalytic fluorescence system were investigated. As shown in Fig. 2b, the kinetic curve revealed that OPD oxidation by H2O2 generated weak fluorescence; whereas in the presence of HRP, fluorescence emission of the system could be much enhanced under the same conditions. Thus, the results demonstrated that OPD oxidation was efficiently catalyzed by HRP.

Characterization of the HRP–OPD–H2O2 fluorescence system. a Fluorescence spectra for the HRP–OPD–H2O2 and OPD–H2O2, which showed the emission of fluorescence 580 nm. Conditions: 1 ng/mL HRP, 25 mmol/L OPD, 1.5 mmol/L H2O2. b Kinetic characteristics of the OPD–H2O2, in the presence of HRP and in the absence of HRP. Conditions: 1 ng/mL HRP, 25 mmol/L OPD, 1.5 mmol/L H2O2, ex/em with 398 nm/580 nm. FL fluorescence
Optimization of experimental conditions
Firstly, UV–Vis absorption spectrophotometry was used to characterize the MBs–Ab conjugates, as shown in the Electronic Supplementary Material (ESM) Fig. S1, where the change of UV–Vis absorption spectra before and after incubating McAb with MBs indicated that McAb was successfully conjugated to MBs. Subsequently the experimental conditions were optimized. Generally speaking, the conditions of immuno-reagents and substrates affect the sensitivity of an immunoassay severely. Therefore, the dilution ratios of biotin–PcAb, streptavidin-labeled HRP, the amount of magnetic beads, and catalytic time were optimized to obtain high F/F 0 ratios (F was the fluorescence signal with the samples containing p24, whereas F 0 was obtained for PBS buffer using the same process as the samples containing p24), and concentrations of OPD and H2O2 were optimized to get the highest fluorescence signal.
The effect of the dilution ratio of biotin–PcAb on F/F 0 was examined and optimized. F/F 0 increased when the dilution ratios of the biotin–PcAb changed from 1:400 to 1:800, and then decreased significantly, as shown in Fig. 3a. Low dilution ratios of biotin–PcAb could result in nonspecific adsorption, whereas too high dilution ratios were not enough to cover the IMBs (as shown in ESM Fig. S2a). Thus, a 1:800 dilution ratio of biotin–PcAb was selected for the subsequent experiments.

Effects of experimental conditions on the signals of the system. a Effect of the dilution ratio of biotin–PcAb. Conditions: 22.5 pg/mL p24; 15 μg IMBs; 1:1000 dilution ration of streptavidin-labeled HRP; 25 mmol/L OPD; 1.5 mmol/L H2O2; catalytic time of 10 min. b Effect of dilution ratio of streptavidin-labeled HRP. Conditions: 1:800 dilution ratio of biotin–PcAb; other conditions were the same as those for Fig. 3a. c Effect of IMBs amount. Conditions: 1:800 dilution ratio of biotin–PcAb; 1:2000 dilution ratio of streptavidin-labeled HRP; other conditions were the same as those for Fig. 3a. d Effect of OPD concentration. Conditions: 1:800 dilution ratio of biotin–PcAb; 1:2000 dilution ratio of streptavidin-labeled HRP; 3 μg IMBs; other conditions were the same as those for Fig. 3a. e Effect of H2O2 concentration. Conditions: 1:800 dilution ratio of biotin–PcAb; 1:2000 dilution ratio of streptavidin-labeled HRP; 3 μg IMBs; 25 mmol/L OPD; other conditions were the same as those for Fig. 3a. f Effect of catalytic time. Conditions: 1:800 dilution ratio of biotin–PcAb; 1:2000 dilution ratio of streptavidin-labeled HRP; 3 μg IMBs; 25 mmol/L OPD; 1.5 mmol/L H2O2; other conditions were the same as those for Fig. 3a. Three independent measurements were taken from three individual preparations for each condition. Error bars indicate the standard deviations
The influence of the dilution ratio of streptavidin-labeled HRP was tested from 1:500 to 1:4000. As displayed in Fig. 3b, F/F 0 was enhanced when the dilution ratio of streptavidin-labeled HRP changed from 1:500 to 1:2000. The highest F/F 0 ratio was attained when the dilution ratio was 1:2000. Further increasing the dilution ratio led to unsaturated reaction (as shown in ESM Fig. S2b). The dilution ratio of streptavidin-labeled HRP of 1:2000 was therefore selected in the subsequent assays.
The effect of the amount of IMBs (1–15 μg) on F/F 0 was also explored. As illustrated in Fig. 3c, F/F 0 reached a maximum on the addition of 3 μg IMBs and then decreased. The reason may be that high concentrations of IMBs induced nonspecific absorption of biotin–PcAb and streptavidin-labeled HRP (as shown in ESM Fig. S2c), which led to poorer sensitivity [41]. Since 3 μg IMBs possessed sufficient binding sites and highly efficient capture capacity for the target in the assay system, 3 μg IMBs was added in the subsequent experiments.
The influence of OPD concentration (varying between 15 and 35 mmol/L) on the fluorescence intensity was investigated. As exhibited in Fig. 3d, the fluorescence intensity reached a plateau with OPD concentration up to 25 mmol/L, which ensured complete HRP-mediated reaction. Further addition of OPD did not obviously enhance the fluorescence intensity. Accordingly, 25 mmol/L OPD was then used in the further experiments.
The effect of H2O2 concentration on the fluorescence intensity was investigated by varying the concentration of H2O2 from 0.5 to 2.5 mmol/L. As seen from Fig. 3e, the fluorescence intensity arrived at the maximum value at 1.5 mmol/L H2O2 and then gradually decreased with increasing concentration of H2O2. The reason may be the reduction of H2O2 on the catalytic activity of HRP [42]. Therefore, 1.5 mmol/L was used as the H2O2 concentration in our experiments.
The effect of catalytic time on the F/F 0 was also studied. F and F 0 were both increased with prolonged catalytic time, but the growth rate of F 0 was faster than that of F when the catalytic time was more than 10 min (as shown in ESM Fig. S2d). Figure 3f showed that the highest F/F 0 was obtained using 10 min as catalytic time. On the basis of this finding, a catalytic time of 10 min was selected for subsequent experiments.
Detection of p24 in buffer solution
Under the optimized conditions, p24 standard solutions were prepared and examined. As shown in Fig. 4, the fluorescence emission at 580 nm increased as the concentration of p24 increased. The calibration plots showed a good linear relationship between the fluorescence intensity and the concentration of p24 in the range from 1.4 to 90.0 pg/mL. The regression equation is Y = 198.3X + 865.3, where Y is the fluorescence intensity of reaction solution and X is the p24 concentration (pg/mL). The linear regression equation found a linear regression coefficient of 0.992. The detection limit (3σ, where σ is the standard deviation of a blank solution processed in the same manner as the samples containing p24, n = 9 (data are shown in ESM Table S1) was estimated to be 0.5 pg/mL. In order to test the reproducibility of the proposed method, 1.4 pg/mL, 22.5 pg/mL, and 45.0 pg/mL p24 were detected, and the relative standard deviations (RSD) were 4.4 %, 4.7 %, and 5.0 %, respectively (data were shown in ESM Table S2). The proposed assay had sufficient reproducibility and good reliability for the detection of p24.

Calibration curve for p24 detection in buffer solution. a Fluorescence spectra of immunoassay with target p24 concentration of 1.4, 2.8, 5.6, 11.2, 22.5, 45.0, 90.0 pg/mL (curve a–g, respectively). b Calibration curve of fluorescence intensity along with p24 concentration. Three independent measurements were taken from three individual preparations for each condition. Error bars indicate the standard deviations
The selectivity was investigated by testing the response of the catalytic fluorescent immunoassay to interfering proteins. Pathogenic (human AFP and human CEA) and abundant proteins (HSA) were introduced into the detection system. Figure 5 shows that the fluorescence signal in the presence of p24 could be clearly distinguished from other proteins, which confirmed the high selectivity of this assay as a result of the highly specific recognition between p24 and its antibodies.

Fluorescence signal of the proposed immunoassay in the presence of different proteins. The concentration of AFP and CEA were 100 ng/mL, concentration of HSA was 100 mg/mL, and concentration of p24 was 22.5 pg/mL. Three independent measurements were taken from three individual preparations for each condition. Error bars show the standard deviations
Detection of p24 in serum
The most crucial application of an analytical method is in routine analysis of analytes in a given biological matrix [18]. The p24 assay in commercial human serum was also performed. As shown in Fig. 6, the calibration curve showed good linearity with R 2 = 0.995 and the detection limit was calculated to be 0.8 pg/mL. The standard addition method was introduced to evaluate the reliability of the proposed method by testing human serum samples spiked with different amounts of p24. As shown in Table 1, the recovery percentage of p24 detected by the assay ranged from 94.8 % to 101.8 % for the different amounts of p24 added, which was satisfactory for quantitative assays of biological samples.

Calibration curve of fluorescence intensity along with p24 concentration in serum. Three independent measurements were taken from three individual preparations for each condition. Error bars indicate the standard deviations
Compared with the conventional ELISA kits (LOD = 10–20 pg/mL) [22], the lower detection limit of the present method was significant for narrowing the period in which to detect HIV-1 infection and could be applied to reduce HIV-1 infection through transfusion of HIV-1-positive blood. Furthermore, the catalytic fluorescent immunoassay coupled with MBs allowed all the assay steps to be completed within 90 min, whereas it takes nearly 3 h for the conventional ELISA kits. In addition, the stability of the prepared IMBs was investigated, and the results indicated the good storage stability at 4 °C during 7 days (as displayed in ESM Fig. S3). Furthermore, for future application of point-of-care testing, the assay time could be shortened by applying some newly developed techniques. Because it took a total of 70 min for incubation and catalysis in our method, automation would accelerate the detection. Magnetic beads can be magnetically manipulated using permanent magnets or electromagnets, so many microfluidic magnetic bead-based assays have been reported [43]. We plan to combine this detection system with lab-on-a-chip format/microfluidic format. Owing to its advantages of simplification, low cost, universality, and sensitivity, the suggested platform can be automated with ease for point-of-care testing.
Conclusions
In summary, we developed a rapid and sensitive approach for the detection of p24 using magnetic beads coupled with catalytic fluorescent immunoassay. Owing to the advantages of high-efficiency immunomagnetic separation, biotin–streptavidin (BA) system amplification, and catalytic fluorescent detection, the detection limit for HIV-1 p24 antigen was low (0.5 pg/mL, 3σ). Furthermore, this assay platform circumvented the interference of magnetic beads in fluorescence detection, and has the advantages of high sensitivity, perfect specificity, good reproducibility, and cost-effectiveness. The lower detection limit of p24 indicated that it can be employed as a promising fast screening method for the detection of HIV-1 infections.
References
Sugarman J. HIV cure research: expanding the ethical considerations. Ann Intern Med. 2013;159(7):490–1. doi:10.7326/00030003-4819-159-7-201310010-00694.
Rodrigo MAM, Heger Z, Cernei N, et al. HIV biosensors - the potential of the electrochemical way. Int J Electrochem Sci. 2014;9(7):3449–57.
Pinching AJ, Jeffries DJ, Harris JRW, Swirsky D, Weber JN. HIV and AIDS. Nature. 1990;347(6291):324. doi:10.1038/347324c0.
Hughes A, Corrah T. Human immunodeficiency virus type 2 (HIV-2). Blood Rev. 1990;4(3):158–64. doi:10.1016/0268-960x(90)90043-R.
Rutherford GW, Schwarcz SK, McFarland W. Surveillance for incident HIV infection: new technology and new opportunities. J Acquir Immun Defic Syndr. 2000;25:S115–9. doi:10.1097/00126334-200012152-00005.
Schupbach J. Viral RNA, and p24 antigen as markers of HIV disease and antiretroviral treatment success. Int Arch Allergy Immunol. 2003;132(3):196–209. doi:10.1159/000074552.
Weber B. Screening of HIV infection: role of molecular and immunological assays. Expert Rev Mol Diagn. 2006;6(3):399–411. doi:10.1586/14737159.6.3.399.
Yilmaz G. Diagnosis of HIV infection and laboratory monitoring of its therapy. J Clin Virol. 2001;21(3):187–96. doi:10.1016/S1386-6532(01)00165-2.
Pilcher CD, Christopoulos KA, Golden M. Public health rationale for rapid nucleic acid or p24 antigen tests for HIV. J Infect Dis. 2010;201:S7–15. doi:10.1086/650393.
Patel P, Mackellar D, Simmons P, et al. Detecting acute human immunodeficiency virus infection using 3 different screening immunoassays and nucleic acid amplification testing for human immunodeficiency virus RNA, 2006-2008. Arch Intern Med. 2010;170(1):66–74. doi:10.1001/archinternmed.2009.445
Hopkins M, Hau S, Tiernan C, et al. Comparative performance of the new Aptima HIV-1 Quant Dx assay with three commercial PCR-based HIV-1 RNA quantitation assays. J Clin Virol. 2015;69:56–62. doi:10.1016/j.jcv.2015.05.020.
McFall SM, Wagner RL, Jangam SR, Yamada DH, Hardie D, Kelso DM. A simple and rapid DNA extraction method from whole blood for highly sensitive detection and quantitation of HIV-1 proviral DNA by real-time PCR. J Virol Methods. 2015;214:37–42. doi:10.1016/j.jviromet.2015.01.005.
Gurtler L. Difficulties and strategies of HIV diagnosis. Lancet. 1996;348(9021):176–9. doi:10.1016/S0140-6736(96)01036-7.
Mcrae B, Lange JAM, Ascher MS, et al. Immune response to HIV P24 core protein during the early phases of human immunodeficiency virus infection. AIDS Res Hum Retrovir. 1991;7(8):637–43. doi:10.1089/aid.1991.7.637.
Contreras M, Barbara J. Screening for HIV P24 antigen. Lancet. 1992;339(8790):438–9. doi:10.1016/0140-6736(92)90134-O.
Beelaert G, Fransen K. Evaluation of a rapid and simple fourth-generation HIV screening assay for qualitative detection of HIV p24 antigen and/or antibodies to HIV-1 and HIV-2. J Virol Methods. 2010;168(1-2):218–22. doi:10.1016/j.jviromet.2010.06.002.
Zhao PX, Feng J, Wu YN, et al. Detection of HIV-1 p24 antigen using a simple and highly sensitive chemiluminescence immunoassay. Futur Virol. 2013;8(7):717–23. doi:10.2217/Fvl.13.45.
Kheiri F, Sabzi RE, Jannatdoust E, Shojaeefar E, Sedghi H. A novel amperometric immunosensor based on acetone-extracted propolis for the detection of the HIV-1 p24 antigen. Biosens Bioelectron. 2011;26(11):4457–63. doi:10.1016/j.bios.2011.05.002.
Fang YS, Huang XJ, Wang LS, Wang JF. An enhanced sensitive electrochemical immunosensor based on efficient encapsulation of enzyme in silica matrix for the detection of human immunodeficiency virus p24. Biosens Bioelectron. 2015;64:324–32. doi:10.1016/j.bios.2014.09.022.
Gan N, Du XW, Cao YT, Hu FT, Li TH, Jiang QL. An ultrasensitive electrochemical immunosensor for HIV p24 based on Fe3O4@SiO2 nanomagnetic probes and nanogold colloid-labeled enzyme-antibody copolymer as signal tag. Materials. 2013;6(4):1255–69. doi:10.3390/Ma6041255.
de la Rica R, Stevens MM. Plasmonic ELISA for the ultrasensitive detection of disease biomarkers with the naked eye. Nat Nanotechnol. 2012;7(12):821–4. doi:10.1038/nnano.2012.186.
Tang SX, Hewlett I. Nanoparticle-based immunoassays for sensitive and early detection of HIV-1 capsid (p24) antigen. J Infect Dis. 2010;201:S59–64. doi:10.1086/650386.
He Q, Zhu ZL, Jin LL, Peng L, Guo W, Hu SH. Detection of HIV-1 p24 antigen using streptavidin-biotin and gold nanoparticles based immunoassay by inductively coupled plasma mass spectrometry. J Anal At Spectrom. 2014;29(8):1477–82. doi:10.1039/C4ja00026a.
Haukanes BI, Kvam C. Application of magnetic beads in bioassays. Bio-Technol. 1993;11(1):60–3. doi:10.1038/Nbt0193-60.
Zhang Y, Zhou DJ. Magnetic particle-based ultrasensitive biosensors for diagnostics. Expert Rev Mol Diagn. 2012;12(6):565–71. doi:10.1586/Erm.12.54.
Stejskalova E, Horakova P, Vacek J, Bowater RP, Fojta M. Enzyme-linked electrochemical DNA ligation assay using magnetic beads. Anal Bioanal Chem. 2014;406(17):4129–36. doi:10.1007/s00216-014-7811-y.
Bettazzi F, Hamid-Asl E, Esposito CL, et al. Electrochemical detection of miRNA-222 by use of a magnetic bead-based bioassay. Anal Bioanal Chem. 2013;405(2-3):1025–34. doi:10.1007/s00216-012-6476-7.
Li YH, Wu ZJ, Liu ZH. An immune sandwich assay of carcinoembryonic antigen based on the joint use of upconversion phosphors and magnetic beads. Analyst. 2015;140(12):4083–8. doi:10.1039/C5an00357a.
Hu Y, Shen GQ, Zhu HL, Jiang GX. A class-specific enzyme-linked immunosorbent assay based on magnetic particles for multiresidue organophosphorus pesticides. J Agric Food Chem. 2010;58(5):2801–6. doi:10.1021/jf903897k.
Wang DB, Tian B, Zhang ZP, et al. Rapid detection of Bacillus anthracis spores using a super-paramagnetic lateral-flow immunological detection system. Biosens Bioelectron. 2013;42:661–7. doi:10.1016/j.bios.2012.10.088.
Malashikhina N, Garai-Ibabe G, Pavlov V. Unconventional application of conventional enzymatic substrate: first fluorogenic immunoassay based on enzymatic formation of quantum dots. Anal Chem. 2013;85(14):6866–70. doi:10.1021/Ac4011342.
Kim YH, Lim HB. Laser-induced fluorescence reader with a turbidimetric system for sandwich-type immunoassay using nanoparticles. Anal Chim Acta. 2015;883:32–6. doi:10.1016/j.aca.2015.04.031.
Bobrow MN, Harris TD, Shaughnessy KJ, Litt GJ. Catalyzed reporter deposition, a novel method of signal amplification - application to immunoassays. J Immunol Methods. 1989;125(1-2):279–85. doi:10.1016/0022-1759(89)90104-X.
Li YX, Hong M, Qiu B, et al. Highly sensitive fluorescent immunosensor for detection of influenza virus based on Ag autocatalysis. Biosens Bioelectron. 2014;54:358–64. doi:10.1016/j.bios.2013.10.045.
Zhu XS, Duan DY, Publicover NG. Magnetic bead based assay for C-reactive protein using quantum-dot fluorescence labeling and immunoaffinity separation. Analyst. 2010;135(2):381–9. doi:10.1039/B918623a.
Kwon Y, Hara CA, Knize MG, et al. Magnetic bead based immunoassay for autonomous detection of toxins. Anal Chem. 2008;80(22):8416–23. doi:10.1021/ac8010044.
Kim JI, Wang CH, Kuizon S, et al. Simple and specific detection of abnormal prion protein by a magnetic bead-based immunoassay coupled with laser-induced fluorescence spectrofluorometry. J Neuroimmunol. 2005;158(1-2):112–9. doi:10.1016/j.jneuroim.2004.08.016.
Berglund GI, Carlsson GH, Smith AT, Szoke H, Henriksen A, Hajdu J. The catalytic pathway of horseradish peroxidase at high resolution. Nature. 2002;417(6887):463–8. doi:10.1038/417463a.
Zhu YD, Peng J, Jiang LP, Zhu JJ. Fluorescent immunosensor based on CuS nanoparticles for sensitive detection of cancer biomarker. Analyst. 2014;139(3):649–55. doi:10.1039/C3an01987j.
Zhag Y, Tan C, Fei RH, et al. Sensitive chemiluminescence immunoassay for E. coli O157:H7 detection with signal dual-amplification using glucose oxidase and laccase. Anal Chem. 2014;86(2):1115–22. doi:10.1021/Ac4028774.
Wang YK, Wang YC, Wang HA, Ji WH, Sun JH, Yan YX. An immunomagnetic-bead-based enzyme-linked immunosorbent assay for sensitive quantification of fumonisin B1. Food Control. 2014;40:41–5. doi:10.1016/j.foodcont.2013.11.025.
Schmidt A, Schumacher JT, Reichelt J, Hecht HJ, Bilitewski U. Mechanistic and molecular investigations on stabilization of horseradish peroxidase C. Anal Chem. 2002;74(13):3037–45. doi:10.1021/ac0108111.
Tekin HC, Gijs MAM. Ultrasensitive protein detection: a case for microfluidic magnetic bead-based assays. Lab Chip. 2013;13(24):4711–39. doi:10.1039/c3lc50477h.
Acknowledgments
We thank Prof. Bing Yan (Wuhan Institute of Virology, Chinese Academy of Sciences) for generous gifts of HIV-1 recombinant p24, biotin-rabbit anti-p24 PcAb, and mouse anti-p24 McAb. We are grateful for the financial support from the Chinese Academy of Sciences (CXJJ-15Z012) and Wuhan Key Laboratory on Emerging Infectious Diseases and Biosafety in Wuhan.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 268 kb)
Rights and permissions
About this article

Cite this article
Zhang, Y., Yang, H., Yu, J. et al. Rapid and sensitive detection of HIV-1 p24 antigen by immunomagnetic separation coupled with catalytic fluorescent immunoassay. Anal Bioanal Chem 408, 6115–6121 (2016). https://doi.org/10.1007/s00216-016-9722-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9722-6