
Abstract
Owing to the high throughput and low cost, next generation sequencing has attracted much attention for SNP genotyping application for researchers. Here, we introduce a new method based on three-round multiplex PCR to precisely genotype SNPs with next generation sequencing. This method can as much as possible consume the equivalent amount of each pair of specific primers to largely eliminate the amplification discrepancy between different loci. After the PCR amplification, the products can be directly subjected to next generation sequencing platform. We simultaneously amplified 37 SNP loci of 757 samples and sequenced all amplicons on ion torrent PGM platform; 90.5 % of the target SNP loci were accurately genotyped (at least 15×) and 90.4 % amplicons had uniform coverage with a variation less than 50-fold. Ligase detection reaction (LDR) was performed to genotype the 19 SNP loci (as part of the 37 SNP loci) with 91 samples randomly selected from the 757 samples, and 99.5 % genotyping data were consistent with the next generation sequencing results. Our results demonstrate that three-round PCR coupled with next generation sequencing is an efficient and economical genotyping approach.

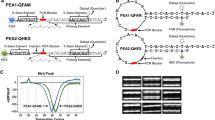
The schematic diagram of three-round PCR
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
SNPs are one of the most important molecular markers that could be associated with complex diseases, and variation of human response to agents and the environment [1, 2]. SNP genotyping is often used in a variety of biological analysis, such as genetic structure analysis, personalized medicine and positional cloning of disease alleles [3, 4]. Thanks to next generation sequencing, the reference genome sequence data and SNP data of different organisms are increasing rapidly in recent years. Therefore, there is an ever-increasing need for SNP genotyping for various purposes [5, 6]. The traditional methods, such as Taqman, Sequenom and SNaPshot [3], are flexible for limited SNP loci for hundreds of samples, but we still need more efficient and economical methods for high throughput SNP genotyping. The SNP genotyping on microarray is a high throughput method which was restricted to designed SNP loci, and are not flexible. Next generation sequencing can provide low cost and high throughput sequencing. This method can simultaneously sequence hundreds of samples to produce thousands or even millions of accurate sequences for genetic variants analysis [7, 8]. Thus next generation sequencing is an emerging high throughput method which is capable of discovering, and genotyping large amounts of SNPs in a single run [7, 9, 10].
A variety of strategies have been integrating next generation sequencing for identification of SNPs and mutations by resequencing. The Current approaches include short PCR [11], long PCR [12], multiplex PCR [13], Hybrid Capture [14, 15] and Molecular Inversion Probes [16]. These strategies are also suitable for SNP genotyping of the known loci. Among them, multiplex PCR is the desirable choice to genotype a large set of SNPs, because it is labor and cost effective, quite flexible to design, and requiring smaller amount of template DNA [8]. However, there still remains two major challenges to perform multiplex PCR coupled with next generation sequencing. The first challenge lies in the difficulty to have uniform abundance of amplicons in one PCR reaction which contains dozens of primer pairs. Different primers have poor uniformity in multiplex PCR because of amplification bias [17], and multiplex PCR is typically limited to simultaneous amplification of 10-20 loci due to primer-dimer formation and mispriming events. In order to reduce the primer competition, researchers used emulsion PCR to separate each pair of primers, while 90.2 % of all amplicons fell within a 25-fold abundance range [18]. The second challenge is the uniformity of products between different samples [11]. Because the multiplex PCR products from thousands samples must be mixed before they are sequenced in next generation sequencing platform, the variations among the samples could cause sequence coverage bias and reduce the sequence efficiency.
Here we present a novel three-round multiplex PCR method coupled to next generation sequencing, which allows for simultaneous amplification of many loci and accurate SNP genotyping. The method enriches target regions in the first two rounds of PCR reaction and as much as possible consume the residual primers to reduce the discrepancy in amplification between different loci. In the third round PCR reaction, the unique adapter primers are added to the ends of target regions of each sample in order to distinguish the SNP information between different samples in the subsequent sequencing reaction. We amplified the 37 loci from 757 human genomic DNA and sequenced the amplicons in a single run on Ion torrent PGM platform. This method has desirable results in terms of coverage and uniformity for the SNP loci after three-round of amplification. We demonstrate that three-round PCR method is multiplex, specific and uniform, which therefore has a huge potential for SNP genotyping using next generation sequencing.
Materials and methods
Primer design
The sequences of 37 targeted regions containing SNP loci were downloaded from the National Center for Biotechnology Information RefSNP database [see Electronic Supplementary Material (ESM) Table S1]. To obtain specific PCR products, we designed chimeric specific primers which contain target sequences and universal sequences (Fig. 1). The product sizes of PCR reaction were between 107 and 160 bp, and the primer length was 37-38 bp, with melting temperature (Tm) 55-65 °C and the GC content between 20 % and 80 %. To distinguish different samples, we designed 40 pairs of adapter primers containing Ion Torrent primer, index sequences and universal sequences (ESM Table S2). Each adapter primer has unique index sequences and represents a non-repeat sample. Therefore, we can simultaneously test 1600 samples using 40 pairs of adapter primers(synthesized by HanYuBio,Shanghai,China). Adapter primers were purified by HPLC, and the other primers were supplied as standard desalting grade.

Schematic overview of primers design features. Each specific primer consists of universal 18 bp flanking sequences (black), unique 19-20 bp targeted arms (red). Specific primer is designed for enrichment of targets containing SNP locus. Adapter primer includes 30 bp ion torrent primer A (pink) which is the sequencing primer or 23 bp ion torrent primer P (green) which is fixed in the bead, unique 10-12 bp index (yellow) and universal sequences (black). The adapter primers add ion torrent primer on the specific primers products in order to can directly put the PCR products in the subsequent emulsion PCR
Three-round PCR
To assess the ability of three-round PCR for SNP genotyping, 757 human genomic DNA (25 ng/ul) from WuXi Mental Health Center were used for genotyping. The method contains three-round PCR. Targets were initially amplified by 10ul PCR reaction containing 2ul human genomic DNA, 1 U of Hot Start DNA Polymerase (Rendu biotechnology, Shanghai, China),1 × PCR buffer (Rendu biotechnology, Shanghai, China), 200 μM dNTPs, 10 mM MgSO4 and 0.05 μM each specific primer in the first round PCR. The following cycling programs were used for the PCR: 94 °C for 15 min, 20 cycles of [94 °C for 30 s, 60 °C for 1 min,72 °C for 30s]. The second round 10ul PCR reaction used 3ul template which is the first round PCR products, 1 U of Hot Start DNA Polymerase (Rendu biotechnology, Shanghai, China),1 × PCR buffer(Rendu biotechnology, Shanghai, China), 200 μM dNTPs, 10 mM MgSO4. The following programs were used for the PCR: 94 °C for 15 min,20 cycles of [94 °C for 30 s, 60 °C for 1 min,72 °C for 30s]. The third round PCR was to add into the second round PCR tube a 10ul reaction mixture which contained 1 U of Hot Start DNA Polymerase (Rendu biotechnology, Shanghai, China), 1 × PCR buffer (Rendu biotechnology, Shanghai, China), 200 μM dNTPs, 10 mM MgSO4 and 0.5 μM mix primers of adapter primer A and adapter primer P. The following programs were used for the PCR: 94 °C for 15 min, 15 cycles of [94 °C for 30 s, 60 °C for 1 min 30s,72 °C for 30s],72 °C for 10 min.
The PCR products of 757 samples were mixed in a 50 ml centrifuge tube after three-round PCR, then the tube was sealed by parafilm and mixed over night. This mixture was purified by TIANgel Midi Purification Kit(TIANGEN BIOTECH, Beijing, China).
Ion torrent PGM sequencing
Purified PCR products mix was then processed for the PGM sequencing process according to the commercially available protocols. PCR products mix was processed on a OneTouch 2 instrument and enriched on a OneTouch 2 ES station. After enrichment, products were sequenced on a 318 chip using the Ion Torrent PGM and the Ion PGMTM Sequencing 200 Kit v2 according to the manufacturer’s instructions.
Sequencing Data Analysis and SNP calling
Usually raw reads including three parts: index sequence, adapter sequence and target sequence. All the sequencing reads were separated according to the corresponding samples based on index sequences by using FASTX-Toolkit with parameter that mismatch base of index sequence was less than 1. After that, the index and adapter sequence were trimed out by using cutadapt software, generating target sequences for each sample. SNPs were identified by using software of BWA (v0.7.12) and samtools (v0.1.19). In brief, target sequences were mapped to the SNP reference sequences (NCBI, dbSNP build 142) to generate sam file by BWA. By using samtools, the sam file was transferred to mpileup file, which was used fot the statistics of SNP bases. For SNP calling, the SNP locus with <15 × coverage were filtered out and heterozygote ratio was fell in 20-80 % range by using in-house perl [19–22].
Results
Overview of three-round PCR
We designed three-round PCR to increase the uniformity of amplicons. In the first round PCR, we use low concentration of specific primers and performed a few cycles. The specific primers are annealed to the target regions and amplified the target sequence (Fig. 1). In the second round PCR, part of products from the first round are added in an new PCR reaction system without adding primers, in order to consume the free residual primers thoroughly from the first round which results in uniform product quantity in multiplex reaction system. In the third round PCR, the target amplicons of each samples are amplified together simultaneously with the adapter primers. Many target loci can be performed in a single tube per sample. To pool and sequence multiple samples, each sample is performed separately and has different adapter primers with unique index sequences to indicate sample identity (ESM Table S3).
Coverage uniformity for amplicons and samples
Uniformity of sequence coverage among targeted amplicons is an important performance metrics because it determines the average depth of amplicons optimal for SNP calling. The purpose of three-round PCR is to reduce the variation in amplification abundance between different loci and samples. All the amplicons of 757 samples were sequenced on Ion torrent PGM platform and the reads of amplicons were discarded if the index sequence had more than one mismatch. We got 5,806,639 raw reads and 4,370,218 reads contained both indexes. Of the reads which contained both indexes, 3,477, 305 reads were usable data and 892,913 reads were the non-target which were discarded. Based on the usable data which had a Phred-like consensus quality ≥ 20, 98.6 % (27613 of 28009) of amplicons were covered at least 1× and 90.5 % (25355 of 28009) of amplicons were covered at least 15×. Among the targets that we sequenced, abundances varied over 1–2 logs (base 10) (Fig. 2).

The coverage range of the amplicons. Amplicons from 757 samples were sequenced on an Ion torrent 318 chip.The x-axis is the number of amplicons which were sequenced least 1× and the y-axis is the number of reads.The logs (base 10) of the estimated relative abundances were calculated. 98.6 % (27613 of 28009) of target amplicons were sequenced one or more times and their abundances varied over 1–2 logs.
We normalized the coverage of each amplicon by the mean coverage (range of reads from 1 to 1657, median reads at 126) of all amplicons, therefore we could directly compare their coverage distribution plots (Fig. 3A). 90.4 % of the SNP loci had reads between 15 and 750, within a 50-fold abundance range (Fig. 3A). The distribution of coverage results showed that 84.5 % of the amplicons was in the range from 15 to 300 reads. The distribution showed that excessively high read depth was around 20 % of all sequence reads (Fig. 4A).

Normalized coverage distribution plots. (A) Shown is the fraction of amplicons (range of reads from 1 to 1657, median reads at 126). (B) Shown is the fraction of samples (range of reads from 25 to 16,520, median reads at 4,593).The normalized coverage was obtained by dividing the observed coverage by the mean coverage.The x-axis is the average normalized coverage and the y-axis is cumulative distribution

Distribution of amplicons. (A) The reads distribution of amplicons.The x-axis is depth of amplicons which were sequenced at least 1×. The y-axis is the number of amplicons. While the most reads (84.5 %) was concentrated within low read depth amplicons (range of reads from 15 to 300). (B) Shown is the total reads of each SNP locus.The logs (base 10) of the estimated relative abundances were calculated in figure
Similarly, we normalized the coverage of each sample (range of reads from 25 to 16,520, median reads at 4,593) (ESM Fig. S1).The data showed that 98.4 %, 97.9 %, 94.3 % and 68.8 % samples fell within 30-fold, 20-fold, 10-fold and 5-fold range (Fig. 3B). At the same time, each SNP locus total reads of 757 samples was different within 12 times (Fig. 4B). For the 37 loci, 31 loci were genotyped in ≥87 % of the individual samples and the other 6 loci were genotyped in <87 % of the total samples. (ESM Table S4).
Accuracy of SNP genotyping
To evaluate the accuracy of genotyping by three-round PCR and next generation sequencing, we compared genotyping data obtained from our new method with data from a conventional SNP genotyping approach named ligase detection reaction (ligase detection reaction, LDR). LDR is a high accuracy method for SNP genotyping. This method uses probes to detect the SNP locus through a ligase, whereas a mismatch at the junction inhibits ligation [3]. 19 SNP loci of 91 samples analyzed by the three-round PCR and next generation sequencing and were reanalyzed using LDR approach (ESM Table S5). The data of next generation sequencing with amplicon reads ≥15, were used for subsequent analysis (ESM Table S6). At the same time, We used a standard procedure for SNP calling in which the probability of a heterozygous individual was falling in the 20-80 % range [19, 20, 22]. 99.5 % genotyping data of LDR approach were consistent with the next generation sequencing results (ESM Tables S5 and S6).
In order to evaluate the allelic bias of next generation sequencing data, we focused on allelic frequencies of next generation sequencing data which was demonstrated by LDR. The homozygous positions had an allelic ratio of ≤0.1 or ≥0.9 and the heterozygous variants showed a ideal distribution converging to 0.5 with increasing coverage (Fig. 5).

Allelic bias of overlapping positions. There are 1682 positions within next generation sequencing data (plotted points in the figure) which are overlapped with LDR results and have 15 times coverage at least.Colors correspond to genotypes, Red, homozygous,black, heterozygous
Discussion
Next generation sequencing is a powerful tool to study genetic variations and mutations. SNP genotyping followed by multiplex PCR is an emerging application of next generation sequencing. Now, more and more suppliers including Illumina and Thermo-Fisher provide commercial kits, such as Illumina TruSeq Amplicon CancerPanel(212 amplicons) and Ion AmpliSeq™ Comprehensive Cancer Panel (16,000 amplicons) [23, 24]. At the same time, there are emerging flexible and non-commercial methods for multiple samples SNP genotyping with next generation sequencing.Three-round PCR is similar to these non-commercial technologies, such as High-plex PCR and GT-seq, which is most suitable for large number of samples [8, 25]. To compare these non-commercial technologies, the uniformity of multiplex PCR is the most important parameters[18]. We amplified 37 SNP loci from 757 DNA samples simultaneously and 90.4 % of amplicons fell within 50-fold. The results indicated that three-round PCR had uniformity of coverage similar to these methods as listed (Table 1).
There were 5,806,639 raw reads and 4,370,218 reads contained both indexes. Of the reads which contained both indexes, 3,477, 305 reads were usable data. 80 % usable data focused on low read depth amplicons (84.5 % amplicons range from 15 to 300 reads) to ensure that most amplicons had enough reads for a high quality genotype assignment. The reads distribution from our results supports that SNP genotyping as described in our method does not require the increase of excessively high sequencing depth to reach reads threshold, therefore it is more cost effective [26].
Among the usable data, 90.5 % amplicons were covered at least 15× and 90.4 % amplicons had reads between 15 and 750 within a 50-fold abundance range. At the same time, 31 loci of the 37 loci were genotyped in ≥87 % of all the individual samples and the other 6 loci were genotyped in ≥40 % of the total tested samples.The results were similar to the previously published results using GT-seq [8].
For many genetic studies it is necessary to examine large numbers of samples for SNP genotyping. This strategy can reduce the cost of oligonucleotides synthesis and the input of template DNA. The experimental results showed that 98.4 % samples reads fell within 30-fold range. This result was due to the fact that the different index sequences had different amplification efficiency [12].
The traditional multiplex PCR methods for target regions are always two-round PCR [17, 18]. The target regions are amplified in the first round PCR reaction, then the adapter primers are added to the second round PCR reaction [25]. These methods always need expensive hardware conditions [18] or many hands-on steps which involve enzymatic processing to reduce the discrepancy in amplification efficiency between different loci [17]. However, three-round PCR only need standardized experimental conditions to do the same work of these methods. Our method uses first two rounds PCR reaction to amplify the target regions and the third round PCR to add the adapter primers (Fig. 1). The first two round of the PCR in our approach are set to reduce the verigation of amplification efficiency between different loci, and the third round PCR using universal primers guarantees uniform quantity in PCR products between different samples which are differentiated by the index sequences.
The SNP genotyping accuracy of three-round PCR and next generation sequencing is quite satisfactory. With this method, 99.5 % genotyping data of LDR approach were found consistent with the next generation sequencing results from the new method. The allelic frequencies of the consistent data was consistent with the reference values to ensure that the SNP genotyping becomes simple and efficient (Fig. 5). The method has extensive application possibilities which require high throughput SNP genotyping. Importantly, this method may not only limited to SNPs genotyping, but could be also suitable for whole genome re-sequencing. With three-round PCR, we can easily amplify and sequence the selected candidate regions in large samples to identify variants associated with disease.
Conclusions
We have presented a new multiplex PCR method in combination with the next generation sequencing for SNP genotyping. The method has good uniformity and simultaneously detects hundreds samples in an unprecedentedly efficient manner, and such a method can be applied for genetic analysis of large samples.
References
Shavrukov Y, Suchecki R, Eliby S, Abugalieva A, Kenebayev S, Langridge P. Application of next-generation sequencing technology to study genetic diversity and identify unique SNP markers in bread wheat from Kazakhstan. BMC Plant Biol. 2014;14:258.
Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19(6):1124–32.
Syvanen AC. Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat Rev Genet. 2001;2(12):930–42.
Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, Zhang Z, Zhang Y, Wang W, Li J, Wei F, Li H, Jian M, Li J, Zang Z, Nielsen R, Li D, Gu W, Yang Z, Xuan Z, Ryder OA, Leung FC, Zhou Y, Cao J, Sun X, Fu Y, Fang X, Guo X, Wang B, Hou R, Shen F, Mu B, Ni P, Lin R, Qian W, Wang G, Yu C, Nie W, Wang J, Wu Z, Liang H, Min J, Wu Q, Cheng S, Ruan J, Wang M, Shi Z, Wen M, Liu B, Ren X, Zheng H, Dong D, Cook K, Shan G, Zhang H, Kosiol C, Xie X, Lu Z, Zheng H, Li Y, Steiner CC, Lam TT, Lin S, Zhang Q, Li G, Tian J, Gong T, Liu H, Zhang D, Fang L, Ye C, Zhang J, Hu W, Xu A, Ren Y, Zhang G, Bruford MW, Li Q, Ma L, Guo Y, An N, Hu Y, Zheng Y, Shi Y, Li Z, Liu Q, Chen Y, Zhao J, Qu N, Zhao S, Tian F, Wang X, Wang H, Xu L, Liu X, Vinar T, Wang Y, Lam TW, Yiu SM, Liu S, Zhang H, Li D, Huang Y, Wang X, Yang G, Jiang Z, Wang J, Qin N, Li L, Li J, Bolund L, Kristiansen K, Wong GK, Olson M, Zhang X, Li S, Yang H, Wang J, Wang J. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463(7279):311–7.
Xu X, Liu X, Ge S, Jensen JD, Hu F, Li X, Dong Y, Gutenkunst RN, Fang L, Huang L, Li J, He W, Zhang G, Zheng X, Zhang F, Li Y, Yu C, Kristiansen K, Zhang X, Wang J, Wright M, McCouch S, Nielsen R, Wang J, Wang W. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 2012;30(1):105–11.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12(7):499–510.
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012:251364.
Campbell NR, Harmon SA, Narum SR. Genotyping-in-Thousands by sequencing (GT-seq): a cost effective SNP genotyping method based on custom amplicon sequencing. Mol Ecol Resour. 2015;15(4):855–67.
Boland JF, Chung CC, Roberson D, Mitchell J, Zhang X, Im KM, He J, Chanock SJ, Yeager M, Dean M. The new sequencer on the block: comparison of Life Technology's Proton sequencer to an Illumina HiSeq for whole-exome sequencing. Hum Genet. 2013;132(10):1153–63.
Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73.
Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–74.
Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, Pawlowski TL, Laub T, Nunn G, Stephan DA, Homer N, Huentelman MJ. Identification of genetic variants using bar-coded multiplexed sequencing. Nat Methods. 2008;5(10):887–93.
Meuzelaar LS, Lancaster O, Pasche JP, Kopal G, Brookes AJ. MegaPlex PCR: a strategy for multiplex amplification. Nat Methods. 2007;4(10):835–7.
Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27(2):182–9.
Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, Weinstock GM, Gibbs RA. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4(11):903–5.
Porreca GJ, Zhang K, Li JB, Xie B, Austin D, Vassallo SL, LeProust EM, Peck BJ, Emig CJ, Dahl F, Gao Y, Church GM, Shendure J. Multiplex amplification of large sets of human exons. Nat Methods. 2007;4(11):931–6.
Varley KE, Mitra RD. Nested Patch PCR enables highly multiplexed mutation discovery in candidate genes. Genome Res. 2008;18(11):1844–50.
Tewhey R, Warner JB, Nakano M, Libby B, Medkova M, David PH, Kotsopoulos SK, Samuels ML, Hutchison JB, Larson JW, Topol EJ, Weiner MP, Harismendy O, Olson J, Link DR, Frazer KA. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat Biotechnol. 2009;27(11):1025–31.
Smith DR, Quinlan AR, Peckham HE, Makowsky K, Tao W, Woolf B, Shen L, Donahue WF, Tusneem N, Stromberg MP, Stewart DA, Zhang L, Ranade SS, Warner JB, Lee CC, Coleman BE, Zhang Z, McLaughlin SF, Malek JA, Sorenson JM, Blanchard AP, Chapman J, Hillman D, Chen F, Rokhsar DS, McKernan KJ, Jeffries TW, Marth GT, Richardson PM. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008;18(10):1638–42.
Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12(6):443–51.
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15(2):121–32.
Hedges DJ, Burges D, Powell E, Almonte C, Huang J, Young S, Boese B, Schmidt M, Pericak-Vance MA, Martin E, Zhang X, Harkins TT, Zuchner S. Exome sequencing of a multigenerational human pedigree. PLoS One. 2009;4(12):e8232.
Sie D, Snijders PJ, Meijer GA, Doeleman MW, van Moorsel MI, van Essen HF, Eijk PP, Grunberg K, van Grieken NC, Thunnissen E, Verheul HM, Smit EF, Ylstra B, Heideman DA. Performance of amplicon-based next generation DNA sequencing for diagnostic gene mutation profiling in oncopathology. Cell Oncol. 2014;37(5):353–61.
Tsongalis GJ, Peterson JD, de Abreu FB, Tunkey CD, Gallagher TL, Strausbaugh LD, Wells WA, Amos C I. Routine use of the Ion Torrent AmpliSeq Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin Chem Lab Med. 2014;52(5):707–14.
Nguyen-Dumont T, Pope BJ, Hammet F, Southey MC, Park DJ. A high-plex PCR approach for massively parallel sequencing. BioTech. 2013;55(2):69–74.
Teer JK, Bonnycastle LL, Chines PS, Hansen NF, Aoyama N, Swift AJ, Abaan HO, Albert TJ. Nisc Comparative Sequencing Program. Margulies EH. Green ED. Collins FS. Mullikin JC. Biesecker LG. Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20(10):1420–31.
Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat Methods. 2009;6(5):315–6.
Acknowledgments
The authors acknowledge support for this work by grants from the National Natural Science Foundation of China (no.31371257) and the Key Project of Science and Technology Commission of Shanghai Municipality (no.14140900502).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 591 kb)
Rights and permissions
About this article

Cite this article
Chen, K., Zhou, Yx., Li, K. et al. A novel three-round multiplex PCR for SNP genotyping with next generation sequencing. Anal Bioanal Chem 408, 4371–4377 (2016). https://doi.org/10.1007/s00216-016-9536-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9536-6