
Abstract
An integrated system was developed for directly processing living cells into peptides of membrane proteins. Living cells were directly injected into the system and cracked in a capillary column by ultrasonic treatment. Owing to hydrophilicity for broken pieces of the cell membrane, the obtained membranes were retained in a well-designed bi-filter. While cytoplasm proteins were eluted from the bi-filter, the membranes were dissolved and protein released by flushing 4 % SDS buffer through the bi-filter. The membrane proteins were subsequently transferred into a micro-reactor and covalently bound in the reactor for purification and digestion. As the system greatly simplified the whole pretreatment processes and minimized both sample loss and contamination, it could be used to analyze the membrane proteome samples of thousand-cell-scales with acceptable reliability and stability. We totally identified 1348 proteins from 5000 HepG2 cells, 615 of which were annotated as membrane proteins. In contrast, with conventional method, only 233 membrane proteins were identified. It is adequately demonstrated that the integrated system shows promising practicability for the membrane proteome analysis of small amount of cells.

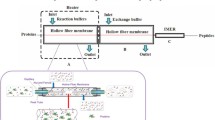
The legend of online abstract figure is (a) schematic illustration of membrane proteins extraction, purification and digestion from living cells; (b) diagrammatic sketch of the automatic integrated membrane proteome analysis system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is well known that membrane proteins (MPs) play a very important role in the occurrence, development, and prognosis of various diseases, especially in tumor [1]. However, the analysis of MPs has always been involved in a series of complex pretreatments and the consequent sample loss. Thus, up to now, the studies of MPs are mostly based on abundant starting materials due to drastic loss during the traditional extraction process. For example, Masuda et al. extracted MPs from 107 Hela cells [2]. However, in clinical applications, it is a vital problem because it is fairly difficult to collect sufficient tissues or cells for detection [3]. Thus, it is more valuable in practical application to develop convenient and effective strategies for profiling MPs with a small amount of samples. To realize the target, one of the most insistent requirements is an automatic and efficient MPs pretreatment method with minimum sample loss and lower sample contamination.
Alternative methods were developed to obtain relatively purified membrane factions. For example, sucrose gradient centrifugation has been used to isolate the inner and outer membranes of rat liver tissue [4]. Aqueous polymer two-phase enrichment strategy has been applied to the rat cerebellum [5, 6]. Some cell surface capture strategies based on electrostatic interaction [7] or specific interactions [8, 9] were developed for acquiring plasma membrane fraction. All of these protocols have achieved great success in the analysis of membrane proteomics, but usually involved buffer exchange, centrifugation, precipitation, or protein modification. Relatively complicated sample pretreatment is not suitable for practical application.
In order to reduce sample loss and contamination, many promising pretreatment approaches have been developed to facilitate the characterization of membrane proteome by integrating the pretreatment steps. Pham et al. [10] established a platform with surface-oxidized nanodiamond particles based on strong hydrophobic forces. Then, the extraction, concentration, purification, and digestion of MPs could be implemented on the particles. Besides, Zhou et al. [11] described a centrifugal proteomic reactor using strong cation exchange (SCX) resin to enrich MPs and replace organic solvent. MPs extraction, digestion, and fractionation could be simply implemented by centrifugation. Whereas the interaction between proteins and materials were based on non-covalent bond, these strategies were inapplicable in the system containing high concentration of sodium dodecyl sulfate (SDS). Thus, some extremely hydrophobic MPs would be lost in the extraction process. In this work, we employed tresyl chemistry to immobilize MPs on the surface of functioned materials. Based on this method, high concentration of SDS could be removed efficiently.
To achieve low-number-cell proteomic analysis, several groups devoted many efforts into setting up methods to minimize the chance of sample transfer. Recently, our group has developed an ultrasensitive approach, an integrated proteome analysis device, iPAD-100 [3, 12]. More than 800 proteins were identified for 100 cell samples. Figeys’ group established a series of microfluidic proteomic reactor based on SCX beads [13, 14]. These strategies showed great superiorities in the rare cell proteomic analysis. Zhang’s group also hammer at the analysis of minute amount samples by online systems [15] and have already made a breakthrough in the membrane proteomic research [16]. They constructed an “SCX-SAX” biphasic capillary column to fulfill membrane protein preconcentration, pH adjustment, reduction, and alkylation, as well as tryptic digestion. This strategy could reduce the starting amount down to 50 ng, and identified 64 integral membrane proteins (IMPs).
On account of these success explorations, online-integrated platform has great application foreground in the analysis of rare cell membrane proteome. In this current work, we firstly established an automatic integrated system for the comprehensive profile of MPs. The system included two core components, a bi-filter and a micro-reactor, which realized the automatic extraction, purification, and digestion of MPs. The experiment procedure design was presented in Fig. 1. Living cells were directly injected into the system and disrupted through ultrasonic treatment. The obtained cell debris was treated by a home-made bi-filter, and divided into two fractions (membrane fraction and cytoplasm fraction) based on size exclusion and hydrophobic properties. MPs were released by high concentration of SDS (4 %). The purification and digestion were implemented in a micro-reactor based on the covalent binding strategy [17, 18]. The whole pretreatment steps were integrated together and carried out automatically. Then, both sample loss and contamination were minimized greatly. According to the experimental results, the proposed system showed excellent performance with good reproducibility and acceptable stability for the identification of MPs from 5000 cells.

Schematic representation of MPs extraction, purification, and digestion from living cells. Step 1, living cells were disrupted in a capillary column by ultrasonic treatment. Step 2, cell debris was transferred in the home-made bi-filter. Membrane fraction was trapped in the bi-filter, while cytoplasm fraction was eluted away. Step 3, MPs were released by washing with extraction buffer, purified by immobilizing on the surface of the packings in a micro-reactor, and digested in the micro-reactor
Experimental
Materials and reagents
TOYOPEARL AF-Epoxy-650M was obtained from TOSOH Bioscience Co. Ltd. (Tokyo, Japan). The protein binding capacity was more than 60 mg per gram dried gel. Silicon dioxide (SiO2) microspheres (∼6 μm) were provided by Nano-Micro Tech (Suzhou, China). PEEK tubes (1/16′′ × 0.03′′) and zero-dead-volume (ZDV) unions were purchased from IDEX Health & Science (Oak Harbor, USA). Then, 6-port 2-position switching valve, 10-port 2-position switching valve, and stainless shims (1/16′′ OD, 0.5 μm) were provided by Valco Instruments Co. Inc. (Houston, USA). All capillary tubes were obtained from Reafine Chromatography Co. Ltd. (Handan, China). EDTA-free Protease Inhibitor Cocktail Tablets was purchased from Roche Applied Science (Indianapolis, IN, USA). All other reagents were domestic products of the highest grade available.
Cell culture and preparation
Human liver hepatocellular carcinoma (HepG2) cell line was provided by Shanghai Medical College of Fudan University (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10 % fetal bovine serum (FBS), 200 U/mL penicillin, and 100 U/mL streptomycin. Cells were harvested by trypsin treatment and washed three times with PBS. The concentration of harvested cells was assayed by flow cytometry (AccuriC6, BD, USA). Cell solution was diluted into 500 cells per microliter with lysis buffer (0.1 M Tris-HCl containing 1 M urea and cocktail, pH 8.0).
Fabrication of bi-filter, micro-reactor, and trapping column
In consideration of the different size of membrane fragments and organelles, we designed a bi-filter with two gap sizes. The bi-filter was fabricated using a short piece of PEEK tube and a stainless shim with the pore size of 0.5 μm as the plunger. Because of the good biocompatibility, SiO2 microspheres with an average size of 6 μm were selected as the packings of the bi-filter. The clearance of the packings (∼2 μm) formed the first filter, and the pore of the stainless plunger formed the second filter. The fabrication procedure in detail was as follows. Prior to packing, PEEK tubes and capillaries were rinsed with methylene chloride (CH2Cl2) and dried by passage of nitrogen gas. Capillaries with large inner diameter and PEEK tubes were installed to ZDV unions with stainless shims. SiO2 microspheres suspended in anhydrous ethanol were firstly packed into a 10-cm long capillary (∼530 μm, i.d.) using a manual pump, and then reversely eluted into the PEEK tube (∼750 μm, i.d.) with well-controlled length (3.5 cm). The nonspecific protein adsorption was considered by evaluating the recovery of both soluble proteins and transmembrane proteins. We took bovine serum albumin (BSA) as the representative of soluble proteins and wide-type bacteriorhodopsin (BR) isolated from Halobacterium salinarum as the representative of transmembrane proteins. Different amount (5 μg and 100 ng) of each protein were respectively injected into the system. Then, lysis buffer (0.1 M Tris-HCl with 1 M urea and cocktail, pH 8.0) and extraction buffer (0.5 M sodium phosphates dissolved in 4 % SDS aqueous solution, pH 8.0) were employed to separately elute BSA and BR. We compared the collected eluates with the original protein solutions by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (for data, see Electronic Supplementary Material Fig S1). According to the intensity results, there is no significant intensity decrease in both bands of BSA and BR eluates. Thus, the nonspecific adsorption could be neglected in the case of soluble proteins or transmembrane proteins. And the amount of proteins did not show any influence to the nonspecific adsorption of the system.
In order to realize the purification and digestion of membrane proteins, a micro-reactor was designed to covalent bind membrane proteins. In our previous work [17], we have emphatically discussed the digestion efficiency of covalent binding method. The results indicated an improvement in the efficiency of tryptic digestion by the covalent binding method. Thus, in this work, we designed a home-made micro-reactor based on the covalent binding method. In addition, we chose a commercial resin to make sure the stability and reproducibility of the micro-reactor. The micro-reactor was made by packing AF-Tresyl-650M resin into a 5-cm-long PEEK tube (∼500 μm, i.d.) using the similar method mentioned in the bi-filter fabrication section. To eliminate the protein digestion efficacy, the same amount of BR (1 μg) was respectively digested by the home-made micro-reactor-assisted method and the widely used filter-aided sample pretreatment (FASP) strategy [19]. In the micro-reactor, BR was bound on the surface of the packings and digested after washing. In the FASP method, high concentration of SDS was removed by buffer exchange through ultrafiltration. Then, proteolytic enzyme was directly added into the ultrafiltration device to digest the remaining proteins. The obtained two peptide samples were analyzed by an Applied Biosystems 5800 proteomics analyzer using CHCA solution (5 mg/mL α-cyano-4-hydroxycinnamic acid and 0.3 mg/mL diammonium citrate 14 in 50 % acetonitrile solution containing 0.1 % TFA) as the matrix (for data, see Electronic Supplementary Material Fig. S2). The two mass spectra are largely similar, except two more tryptic peptides (peeks at 1322.6 and 1838.8) were detected by the micro-reactor-assisted method. It is probably because that sample loss was decreased by the micro-reactor-assisted method.
Trapping column was made by packing C18-boned particles into capillary (∼75 μm, i.d.) with an on-column frit prepared according to our previous work [20]. All of the prepared devices were flushed using a constant flow pump (Shanghai Wufeng Scientific Instrument Company, China) to make sure the microspheres packed compactly.
Fabrication and workflow of the automatic integrated system
Figure 2 illustrates the instrumental setup of online membrane proteome analysis system. The system consists of a programmable 10-syringe pump, a 6-port 2-position valve (valve A), a 10-port 2-position (valve B), a sample loop, and the prepared bi-filter, micro-reactor, and trapping column.

Diagrammatic sketch of the automatic integrated membrane proteome analysis system. The two valves, marked as valve A and valve B, were connected through port 6 (valve A) and port 2 (valve B) and port 5 (valve A) and port 3 (valve B). A capillary (320 μm i.d. × 15 cm) was fit between port 2 and port 4 on valve A as a sample loop. Bi-filter, micro-reactor, and trapping column were, respectively, mounted to port 1 and 4, port 10 and 7, and port 8 on valve B. Valve positions are given for the sample loading (a), membrane fraction filtration (b), membrane proteins extraction and purification (c), and membrane proteins digestion and peptides trapping (d)
As showed in Fig. 2a, valve A was switched in position 1. Then, 10 μL of living cell solution (∼5000 cells in the lysis buffer, 0.1 M Tris-HCl with 1 M urea and cocktail, pH 8.0) was injected into the sample loop. After sonicate treatment for 0.5 h in ice water at 180 W, cells were disrupted into pieces. Then, valve A was switched in position 2, and valve B was switched in position 1 (Fig. 2b). Cell debris was delivered to the home-made bi-filter at a flow rate of 500 nL/min. And, the whole system was put into a column oven with the temperature fixed at 60 °C. Owing to the different solubility and size, membrane fractions were trapped by the SiO2 microspheres, while small organelles were trapped by the stainless shim. After 2 h, the bi-filter was flushed by water at 2 μL/min for 0.5 h. Then, Tris and urea were removed away from the system to avoid the impact on the subsequent operation. In Fig. 2c, valve B was switched to position 2. Extraction buffer (0.5 M sodium phosphates dissolved in 4 % SDS aqueous solution, pH 8.0) was delivered from port 5 at a flow rate of 500 nL/min to dissolve the trapped membrane fraction. Subsequently, the obtained MPs were covalently immobilized on the surface of packings in the micro-reactor by tresyl chemistry. After another 2 h, MPs were nearly released and reacted completely. Then, the micro-reactor was washed at 2 μL/min with 4 % SDS solution, blocking buffer (0.1 M Tris-HCl with 0.5 M NaCl, pH 8.0), and NH4HCO3 buffer (50 mM, pH 8.0) in sequence, to remove the unreacted substances, block residual tresyl groups on the packings, and remove the high concentration of interferents, respectively. In the last step (see Fig. 2d), valve B was switched back to position 1. From port 9, 20 mM DTT in NH4HCO3 buffer (50 mM, pH 8.0) was delivered by the programmable pump at 1 μL/min for 1 h to reduce the immobilized MPs. Straight after that, the oven temperature was turn down to 37 °C. Then, 50 mM IAA in the same buffer was delivered at the same flow rate for 45 min to alkylate the thiol groups produced in the reduction step. After removing the redundant reagents, 100 ng of trypsin dissolved in 50 mM NH4HCO3 buffer (10 ng/μL) was delivered at 200 nL/min into the micro-reactor. After 1 h, the flow rate was increased to 500 nL/min. Then, obtained proteolytic peptides were retained in the trapping column, and the residual materials were washed by water. Finally, trapping column was mounted onto the RPLC-MS/MS system for separation and identification.
Optimization of the extraction conditions
In order to get a more effective separation, we investigate four conditions (washing volume, extraction volume, washing temperature, as well as extraction temperature) by SDS-PAGE test.
First, we sequentially collected cytoplasm fractions into tubes from port 2 with each sample containing 20 μL. According to the intensity of each slice presented in Electronic Supplementary Material Fig. S3, the concentration of cytoplasm proteins gradually reduced with the increase of elution volume. When employing 80 μL of lysis buffer, no obvious protein bands existed in the corresponding slice. Thus, washing by 60 μL of lysis buffer at a flow rate of 0.5 μL/min could achieve the elution of cytoplasm proteins. Similarly, we collected the membrane fractions from port 10. Furthermore, 40 μL of extraction buffer at 0.5 μL/min was sufficient to elute MPs based on the results showed in Electronic Supplementary Material Fig. S3.
Next, we estimated the effect of temperature on the solubility of cytoplasm proteins and membrane proteins. We carried out four parallel experiments with the elution temperature changing from 30 to 60 °C, and collected all of elution buffer for SDS-PAGE test (data see Electronic Supplementary Material Fig. S4). From both Electronic Supplementary Material Fig. S4a and S4b, the intensity of proteins showed a sharply increasing tendency as the temperature raised from 30 to 50 °C. Based on the results, the solubility of cytoplasm proteins and membrane proteins was improved by rising temperature. It was probably because with the increase of temperature, intensive movements of solvent molecules enhanced the probability of the interaction between solvents and proteins. Moreover, compared with primitive proteins, partly denatured proteins with more loose structures possess higher solubility. In addition, for MPs, it was easier to be released from the phospholipid bilayer and eluted away under a relatively high temperature. It is noteworthy that a slightly increasing intensity was observed when temperature changed from 50 to 60 °C. Considering the subsequent covalent binding reaction, we compared the reaction efficacy operated under 50 and 60 °C. We tested the effluents after reaction, and the results were showed in Electronic Supplementary Material Fig. S4c. Under a higher temperature, more free amino groups of MPs exposed outside, and it was easier to react with the active groups on the surface of AF-Tresyl-650M resins. Therefore, the integrated device was fixed in a column oven and the extraction steps were performed at 60 °C.
Identification of cytoplasm proteins and membrane proteins
Cytoplasm proteins collected from port 2 (valve B) and MPs collected from port 10 (valve B) were both identified by widely used filter-aided sample pretreatment (FASP) method [19]. Besides, the collected MPs were also treated by the offline covalent immobilization strategy. Then, 5 mg of AF-Tresyl-650M resins was mixed with MPs. After reacting for 4 h, the coupled materials were treated by the similar method mentioned above, including residual tresyl groups blocking, interference washing, protein reduction, and digestion. All digestion products were lyophilized for the analysis of RPLC-MS/MS.
Preparation of cytoplasm fraction and membrane fraction by differential centrifugation method
The preparation procedure was according to the published literature with subtle modification [21]. In the differential centrifugation method, cell solution was diluted into 100 cells per microliter with lysis buffer (0.1 M Tris-HCl containing 1 M urea and cocktail, pH 8.0). Then, 500 μL prepared cell suspension (containing 50,000 cells) was lysed by a sonicator (QSONICA, USA) in ice bath and centrifuged at 1000 g for 5 min to pellet unbroken cells and nuclei. Crude membrane fraction was recovered by centrifugation at 100,000 g for 1 h and dissolved by 50 mM PBS buffer (pH 8.0) containing 4 % SDS. Both of cytoplasm proteins in the supernatant and MPs were collected for the subsequent identification by FASP method.
Peptides analysis by RPLC-MS/MS
The experiments were performed on an EASY NANO system connected to a linear ion trap–Orbitrap hybrid mass spectrometer equipped with a nano-electrospray ion source (Thermo Scientific, San Jose, CA, USA). Trapped peptides were desorbed and separated on the analytical column (Acclaim PepMap 100 C18, 75 μm × 15 cm, 3 μm, 100 Å) with a 120 min linear gradient from 0 to 45 % ACN with 0.1 % formic acid. The LTQ-Orbitrap instrument was programmed to operate in a data-dependent mode. Survey full-scan MS spectra with one micro scan (m/z 350–1600) were acquired in the Orbitrap instrument with a mass resolution of 60 K. Up to eight most intense ions per scan were fragmented and detected in the linear ion trap. Dynamic exclusion was enabled to minimize repeated sequencing. Peaks selected for fragmentation more than once within 10 s were excluded from selection for 90 s.
Bioinformatics analysis
The mass spectra were searched using the MaxQuant (version 1.5.2.8, Thermo Scientific) based on the Human UniProtKB/Swiss-Prot database (released on Feb. 2014, with 20,265 entries). The first search mass tolerance and main search mass tolerance were fixed as 20 and 4.5 ppm, respectively. Minimal peptide length was set to seven amino acids and a maximum of two missed cleavages. The minimal number of peptides was set to one for protein identifications with false discovery rate (FDR) less than 0.01. In addition, other parameters were set as follows: enzyme, trypsin; fixed modification, carbamidomethylation (C); variable modification, oxidation (M), acetylization (protein N-term), and deamidation (NQ). The subcellular location of these identified proteins was predicted by gene ontology (GO) component and function terms from UniProt Knowledge base (UniProtKB) [22]. The theoretical molecular weight (MW), isoelectric point (pI), and grand average of hydropathy (GRAVY) value of the identified proteins were calculated using the ProtParam software (http://web.expasy.org/protparam) [23]. Predicted transmembrane domains (TMDs) of identified proteins were obtained by using the transmembrane hidden Markov model (TMHMM) algorithm, available at http://www.cbs.dtu.dk/services/TMHMM [24].
Results and discussion
Evaluation of the filtering extraction efficiency
In the extraction process, cell debris was injected into a well-designed bi-filter, and separated into membrane fraction and cytoplasm fraction. The separation was based on the following two aspects. On the one hand, cytoplasm proteins in cells were released and washed away with lysis buffer. While, the membrane fractions were precipitated in the bi-filter because of the poor solubility in the lysis buffer. On the other hand, because of the hydrophobic interaction, membrane debris aggregated together with a larger size. Thus, membranes with large size were retained in the clearance of the SiO2 packings—the first filter, and organelles with relatively small size were blocked by the second filter—stainless shims with a pore size of 0.5 μm (see Fig. 1, step 2). Until we use the extraction buffer to disrupt the structure of lipid bilayer, MPs could be released and eluted from the bi-filter. The optimized extraction conditions had been investigated by a series of contrast experiments (for data, see Electronic Supplementary Material Figs. S3 and S4).
In order to evaluate the MPs extraction efficiency from small amount of cells, we chose differential centrifugation method as the contrast method to make a comparison with the proposed filtering extraction method. Whereas differential centrifugation method involved ultracentrifugation and sample transfer, we increased the initial amount by ten times (50,000 cells). The collected cytoplasm fractions, washing fractions, and membrane fractions obtained both by the integrated system and differential centrifugation method were tested by SDS-PAGE method (for data, see Fig. 3a, slice 1–6). Differential centrifugation method was implemented based on the different particle size and sedimentation velocity of the organelles components [25]. Thus, it is difficult to extract pure membrane fraction, and there are still some protein bands in the washing fraction slice (Fig. 3a, slice 5). The proposed filtering extraction method was not only based on solubility difference in the different buffer solution but also influenced by the synergy of two-stage size exclusion effect and the interaction between SiO2 microsphere and phospholipid bilayer. As shown in Fig. 3a, slice 2, there is almost no proteins in the washing fraction. Therefore, the on-column method greatly reduced the sample loss of both cytoplasm and membrane proteins.

Comparison of membrane proteins extraction effect between the filtering extraction method and the differential centrifugation method. SDS-PAGE test result of cytoplasm proteins (slice 1 and 4), washing fraction (slice 2 and 5), and membrane proteins (slice 3 and 6), respectively, obtained by the two methods (a). Overlap of total identified proteins between cytoplasm fraction and membrane fraction separated based on the differential centrifugation method (b) and the on-column extraction method (c)
To make a further comparison, we employed the FASP method to digest the collected cytoplasm proteins and membrane proteins mentioned above. The obtained peptide samples were analyzed by RPLC-MS/MS. Figure 3b, c respectively presents the identification results. By the differential centrifugation method, we totally identified 396 and 500 proteins from cytoplasm factions and membrane fractions by 3 replicate experiments. Among them, the overlap part accounted for 42.9 % of the total number of identified proteins. In contrast, with the filtering extraction method, the identification results of cytoplasm fraction and membrane fraction increased by 37.9 and 48.0 %, respectively. Nevertheless, the overlap part only accounted for 18.1 %. These results show that cytoplasm proteins and membrane proteins could be effectively separated by the rational-designed bi-filter.
Proteome profiling of MPs
To evaluate the practicability of the automatic integrated system, three parallel identification experiments were performed by liquid chromatography coupled with LTQ-Orbitrap mass spectrometer. Then, 1018, 1032, and 1041 proteins were identified by each run from 5000 cells. The datasets were listed in Electronic Supplementary Material Table S1. The Electronic Supplementary Material Fig. S5 displays the satisfactory overlap of the results from the three runs and indicates the good reproducibility of the strategy. With merging the identification results from triplicate experiments, the number of identified proteins rose to 1348. Among that, 615 proteins were considered as MPs or associated MPs based on TMHMM, GO slim algorithms, and GRAVY scoring [2].
In order to investigate the efficacy of online purification and digestion by the micro-reactor, we compared it with conventional FASP method and offline covalent binding strategy. The identification results were listed in Table 1. There are more than 60 and 80 % increases in both identified proteins and membrane proteins. As shown in Electronic Supplementary Material Fig. S6, a total of 1523 proteins were identified by the three methods. Among them, 501 (32.9 %) proteins were uniquely identified by the online method, including protein P35499 which possesses 21 TMDs. Moreover, in the 615 identified MPs, 256 proteins were classified as integral membrane proteins according to TMHMM algorithm. The number was 70.1 and 106 % higher than the two control methods, respectively. Based on the on-column method, we could identify a higher proportion of IMPs. It must be because that the online method implemented on an integrated device not only reduced sample loss in MPs and proteolytic peptides elution from a small amount of sample but also improve the digestion efficacy of the transmembrane part.
To further analyze the practicability and efficacy of the automatic integrated system in MPs identification, we compared the result with that based on a conventional method (MPs were purified and digested by FASP method after being extracted by differential centrifugation method). Then, 1348 and 500 proteins were respectively identified by the two methods. Correspondingly, 615 (45.6 %) and 233 (46.6 %) proteins were classified as MPs based on the two methods. In addition, 66.4 % of the total proteins as well as 65.7 % of MPs identified by the conventional method overlap with the result obtained by our automatic system (see the Venn diagrams in Fig. 4a). Moreover, we compared the distributions of IMPs obtained by the two methods (see Fig. 4b). Based on our automatic system, a group of 154 proteins (group A) were classified as IMPs. And, a group of 84 proteins (group B) were classified as IMPs by the conventional method. IMPs possessing one to three transmembrane domains (TMDs) occupied similar percentages in the two groups. IMPs possessing four to six TMDs occupied 9.1 % in group A, less than that in group B (17.9 %). IMPs possessing more than seven TMDs occupied 18.2 % in group A, higher than that in group B (10.8 %). Thus, our automatic system showed an advantage in identifying complex IMPs. Furthermore, the distribution of IMPs from Human UniProtKB/Swiss-Prot database was also presented in the same coordinate chart (black bars). With respect to the number of TMDs, the distribution trend of the identified IMPs based on our system was similar to that of the IMPs in the database in general. While the percentage of identified IMPs with seven TMDs was about 65.9 % lower than that of the IMPs in the database. It is probably because that these proteins would not be expressed in the HepG2 cell line, or the low copies in 5000 cells made them almost impossible to be identified by the current mass spectrometry technology. All of the comparison results indicate that our automatic system showed acceptable reliability and obvious superiority than conventional method.

a Overlap of identification results obtained by conventional method (MPs purified and digested by FASP method after being extracted by differential centrifugation method) and our automatic system; b comparison on TMDs distribution of IMPs in Human UniProtKB/Swiss-Prot database (released on Feb. 2014, with 20,265 entries) and that respectively identified by RPLC-MS/MS with the integrated device and conventional method. Black bars refer to the IMPs in the Human UniProtKB/Swiss-Prot database, and red and blue bars refer to the IMPs respectively identified by the two methods
Conclusion
In summary, the automatic integrated system, a combination of a bi-filter and a micro-reactor, was designed for the analysis of the membrane proteome from a small amount of cells. It not only solved the problems involved in the extraction, purification, and digestion of trace amount MPs but also simplified all of the pretreatment process and minimized sample loss that happened in practice. By such a system, the profiling of membrane proteome from 5000 cells was easily achieved. More transmembrane proteins (including the number and the proportion) were identified, which is one of the most important and challenging part in membrane proteome analysis. As far as we know, there is no report of membrane proteome analysis starting directly with as few as thousands-scale cells. Therefore, the automatic integrated device would greatly facilitate the analysis of membrane proteome of small number of cells.
References
Slamon DJ, Clark GM, Wong SG. Science. 1987;235:177–82.
Masuda T, Tomita M, Ishihama Y. J Proteome Res. 2008;7:731–40.
Chen Q, Yan G, Gao M, Zhang X. Anal Bioanal Chem. 2015;407:1027–32.
Lin Y, Liu Y, Li J, Zhao Y, He Q, Han W, et al. Electrophoresis. 2010;31:2705–13.
Schindler J, Lewandrowski U, Sickmann A, Friauf E. J Proteome Res. 2008;7:432–42.
Zhou J, Lin Y, Deng X, Shen J, He Q, Chen P, et al. J Proteome Res. 2008;7:1778–83.
Durr E, Yu J, Krasinska KM, Carver LA, Yates JR, Testa JE, et al. Nat Biotechnol. 2004;22:985–92.
Elia G. Proteomics. 2008;8:4012–24.
Lee D, Park K, Banerjee M, Ha S, Lee T, Suh K, et al. Nature Chem. 2011;3:154–9.
Pham MD, Yu SS, Han C, Chan SI. Anal Chem. 2013;85:6748–55.
Zhou H, Wang F, Wang Y, Ning Z, Hou W, Wright TG et al. Mol Cell Prot. 2011;O111.008425, 1–11.
Chen Q, Yan G, Gao M, Zhang X. Anal Chem. 2015;87:6674–80.
Tian R, Wang S, Elisma F, Li L, Zhou H, Wang L et al. Mol Cell Prot. 2011;M110.007252, 1-15
Tian R, Hoa XD, Lambert JP, Pezacki JP, Veres T, Figeys D. Anal Chem. 2011;83:4095–102.
Ma J, Liu J, Sun L, Gao L, Liang ZL, Zhang Y. Anal Chem. 2009;81:6534–40.
Zhao Q, Liang Y, Yuan H, Sui Z, Wu Q, Liang Z, et al. Anal Chem. 2013;85:8507–12.
Liu Y, Yan G, Gao M, Deng C, Zhang X. Proteomics. 2015;15:3892–900.
Wang J, Gao M, Yan G, Zhang X. Anal Chim Acta. 2015;880:77–83.
Wisniewski JR, Zougman A, Nagaraj N, Mann M. Nat Methods. 2009;6:359–62.
Zhang X, Huang S. J Chromatogr A. 2001;910:13–8.
Yu Y, Xie L, Gunawardena HP, Khatun J, Maier C, Spitzer W, et al. Anal Chem. 2012;84:9008–14.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Nat Genet. 2000;25:25–9.
Kyte J, Doolittle RF. J Mol Biol. 1982;157:105–32.
Krogh A, Larsson B, von Herijne G, Sonnhammer ELL. J Mol Biol. 2001;305:567–80.
Simpson RJ, Connolly LM, Eddes JS, Pereira JJ, Moritz RL, Reid GE. E1ectrophoresis. 2000;21:1707–32.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Project: 21175026), the National High-Tech R&D Program of China (Project: 2012AA020202), and the National Research Programs of China (Project: 2012CB910604, 2013CB911201, and 2012YQ12004409).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Liu, Y., Yan, G., Gao, M. et al. Integrated system for extraction, purification, and digestion of membrane proteins. Anal Bioanal Chem 408, 3495–3502 (2016). https://doi.org/10.1007/s00216-016-9427-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9427-x