
Abstract
We report the coupling of nano-liquid chromatography (nano-LC) with an ambient dielectric barrier discharge ionization (DBDI)-based source. Detection and quantification were carried out by high-resolution mass spectrometry (MS), using an LTQ-Orbitrap in full scan mode. Despite the fact that nano-LC systems are rarely used in food analysis, this coupling was demonstrated to deliver extremely high sensitivity in pesticide analysis, with limits of detection (LODs) as low as 10 pg/mL. In all cases, the limits of quantification (LOQs) were compliant with the current EU regulation. An excellent signal linearity over up to four orders of magnitude was also observed. Therefore, this method can easily compete with conventional GC-(EI)-MS or LC-ESI-MS/MS methods and in some cases outperform them. The method was successfully tested for food sample analysis, with apples and baby food, extracted using the QuEChERS approach. Our results demonstrate an outstanding sensitivity (at femtogram level) and reproducibility of the nano-LC-DBDI coupling, capable of improving routine pesticide analysis. To the best of our knowledge, this is the most sensitive and reproducible plasma-MS-based method for pesticide analysis reported to date.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The analysis of pesticides in environmental and food samples is important to ensure the absence of potential risks to human health. In Europe, the maximum legally allowed concentration level of pesticides in different food/feed matrices is expressed by their maximum residue levels (MRLs), according to the current EU regulation (EC 396/2005). Although a nanogram-per-gram level of sensitivity is easily achieved with current analytical instrumentation, a sub-nanogram-per-milliliter sensitivity is required in some cases, e.g., for surface and drinking water analysis, where the regulation is generally more strict.
A variety of different methods have been used for multi-residue analysis of these compounds [1], among which gas chromatography (GC) and liquid chromatography (LC) are the most frequently used separation techniques and mass spectrometry is the most powerful and comprehensive technique for their detection and identification. When the absence of banned pesticides has to be ensured, highly sensitive methods are preferred, delivering at least picogram-per-gram sensitivities. In this work, we were able to achieve such a high sensitivity by coupling nano-liquid chromatography with dielectric barrier discharge ionization and high-resolution MS. We show ultrasensitive analysis of pesticides at low picogram-per-gram concentration levels without previous sample up-concentration. We analyzed real food samples using the EU Quick Easy Cheap Effective Rugged and Safe (QuEChERS) multi-residue method CEN 15662 [www.cen.eu] as well as water samples.
Nano-LC was first introduced by Karlsson and Novotny in 1988 [2] and has since then mostly been used for proteomics applications. In most cases, the capillary columns have internal diameters between 10 and 100 μm, and therefore, the flow rates employed are in the nanoliter-per-minute range. Compared to non-miniaturized techniques such as high-pressure liquid chromatography (HPLC) and ultra-high-pressure liquid chromatography (UPLC), there are some advantages in the use of nano-LC systems, e.g., increased analyte sensitivity due to less chromatographic dilution (much lower flow rate and column I.D.) and reduction of solvent consumption [3]. On the other hand, the chromatographic separation of small molecules is in most cases worse than that with UPLC, and average chromatographic runs are longer. Moreover, since only a few tens of nanoliters are usually injected into the column, the sensitivity is not enhanced compared to that of UPLC if more sensitive detectors are not used or higher sample volumes are not injected. In nano-LC, the injection of large sample volumes is often called on-column focusing [4], and works by injecting volumes of up to several microliters in cases where the analytes have a higher affinity towards the column stationary phase compared to their solvent. For all these reasons, nano-LC has not been used extensively for the analysis of compounds of environmental interest [3]. In addition, with the exception of proteomics analysis, compounds separated by nano-LC are rarely detected by mass spectrometry; UV spectroscopy is still the preferred detection method [3].
Nano-LC was already employed by Buonasera et al. for pesticide analysis [5], who compared the performance of three different capillary columns for the separation of eight organophosphorus pesticides in baby food, which were then detected by UV. Despite the use of on-column focusing, which was found to increase sensitivity by two orders of magnitude, reported limits of detection (LODs) were merely between 4.4 and 37.5 ng/mL.
The only nano-LC-MS-based approach used for pesticide analysis was reported by Cappiello et al. [6]. These authors used a direct electron ionization interface for the analysis of organophosphorus pesticides in water to directly introduce and nebulize the eluate into the electron impact ionization (EI) source (in vacuum). After solid-phase extraction (SPE), reported LODs were around 3 pg/mL for four of them. Analysis of environmental samples was also performed with this technique by the same research group [7]. Zhu et al. [8] used a nano-LC system based on a microfluidic chip and an enrichment column to analyze illicit drugs and metabolites in human hair with tandem mass spectrometry, with limits of detection in the low picogram-per-milligram level of hair sample. A multi-walled carbon nanotube-dispersive solid-phase extraction was also used [9] for the extraction of 12 pesticides prior to nano-LC-UV analysis. In that work, a trapping column was employed to increase the overall sensitivity.
Despite the sensitivity of UV detection, MS is the preferred detector in pesticide analysis, due to its sensitivity and specificity, especially in combination with electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI). More recently, different plasma-based ionization sources, including the low-temperature plasma (LTP) source, have been developed and employed for pesticide analysis, where they can deliver very high sensitivity [10–12]. Most of these sources are based on a dielectric barrier discharge, and they are gaining attention in the mass spectrometric community [13].
A comparison of the performances of ESI, APCI, and a helium plasma dielectric barrier discharge ionization (DBDI) source for pesticide analysis was reported by Gilbert-López et al. [14], with the separation of 43 pesticides by LC and vaporization of the eluent (200 μL/min) by a modified commercial Ion MaxTM API source [15] (Thermo Fischer Scientific). This low-temperature microplasma was already used in a similar configuration in combination with liquid chromatography for the analysis of environmental and food samples [14, 16]. This helium plasma ionization source showed low to moderate matrix effects in the 1–200-ng/mL concentration range, with limits of quantification (LOQs) in the low nanogram-per-milligram range, compliant with EU regulations in most cases. However, a significant amount of fragmentation for some molecular species was observed, leading to the conclusion that the ionization conditions of DBDI were less gentle than those of ESI in their setup [14]. Moreover, the presence of many salt adducts could decrease the reproducibility of that method if salt impurities are present in the mobile phase.
Despite the fact that DBDI sources are increasingly widespread, an important question still remains unanswered: could these sources deliver reproducible results over extended periods of time? As most of the DBDI sources are used for ambient mass spectrometry (i.e., no or minimal sample preparation and generation of ions in the ambient environment), the sampling of the ions from the source is a crucial aspect, as such sources are known to be strongly affected by geometric parameters. In our setup, we employ a highly efficient ion sampling technique, which we call active capillary sampling, where the ionization does not happen in an open environment like in ESI, APCI, or conventional LTP sources but inside the source which is constructed as extension of the MS inlet itself. This dramatically increases robustness and ion transmission into the MS.
In this study, we report the direct coupling of a liquid chromatographic system with the active capillary plasma ionization source developed in our laboratory [17] and recently employed for ultrasensitive ambient detection of chemical warfare agents [18]. It is the first reported interfacing of a nano-LC with an ambient DBDI source. As first application for this coupling, we present the ultrasensitive analysis of pesticides in food samples.
Materials
Formic acid, HPLC-grade water, and acetonitrile were obtained from Sigma-Aldrich (Buchs, Switzerland). QuEChERS citrate extraction tubes were obtained from Supelco (Bellefonte, PA, USA; product number 55227-U). PESTANAL© analytical-grade standards were purchased from Sigma-Aldrich and were simazine (CAS 122-34-9), metolcarb (CAS 1129-41-5), dichlorvos (CAS 62-73-7), propoxur (CAS 114-26-1), carbofuran (CAS 1563-66-2), bendiocarb (CAS 22781-23-3), atrazine (CAS 1912-24-9), metalaxyl (CAS 57837-19-1), isoproturon (CAS 34123-59-6), ethiofencarb (CAS 29973-13-5), isoprocarb (CAS 2631-40-5), propham (CAS 122-42-9), terbuthylazine (CAS 5915-41-3), metolachlor (CAS 51218-45-2), malathion (CAS 121-75-5), phoxim (CAS 14816-18-3), imidacloprid (CAS 138261-41-3), and 2,4,5-trichlorophenoxyacetic acid (CAS 93-76-5). Food samples (organic apples and organic apple puree baby food) were purchased at a local grocery store in Zurich.
Sample preparation
Individual pesticide standard stock solutions were prepared at a concentration of 500 μg/mL in acetonitrile and stored at −20 °C. A mix of the pesticides at 20 μg/mL in acetonitrile was prepared from the stock solutions. Diluted solutions used for LC analysis were prepared in H2O/ACN 9:1 (v/v) from the mix and were stored at a temperature of 6 °C. Water dilutions were prepared with HPLC-grade water.
Apples (including peel) were finely homogenized using a blender. A 10.0-g aliquot of the homogenized apple or apple puree baby food was weighted in a 50-mL polypropylene centrifugation tube. Extractions were performed with the EU QuEChERS method, as described in the literature (method CEN 15662), but without the dispersive-SPE cleanup step. After extraction, the acetonitrile supernatant was filtered through a 0.2-μm Teflon filter, transferred into a 20-mL brown vial, and stored at 4 °C before use.
Instrumentation
A lab-built vaporization system allowed for a complete and fast vaporization of the eluent (800 nL/min), an essential requirement to avoid peak tailing and non-quantitative results. For optimizing the position of the capillary inside the vaporization chamber, the vaporization temperature, and the carrier gas flow rate, separate pesticide working standard solutions (100 μg/mL in acetonitrile) were prepared from stock solutions and directly infused into the vaporization chamber by means of a pressure-assisted vaporization system. Thereby, the sample was delivered through a fused silica capillary (I.D. 30 μm, O.D. 365 μm, length 25 cm) connected to a pressurized reservoir to the vaporizer system, mimicking a nano-LC.
The vaporization chamber itself consists of a hollow 50-W heating cartridge (Progab AG) with a 6-mm-diameter bore and a length of 120 mm held at 260 °C by a P.I.D. controller. The fused silica capillary was connected to the chamber by a T-piece and held in place by a PTFE ferrule. The carrier gas was introduced into the chamber using the other connection on the T-junction, resulting in a concentric flow of preheated nitrogen carrier gas around the (nano-LC) capillary. The carrier gas flow was maintained constant at 3 L/min by means of a mass-flow controller (Bronkhorst High Tech B.V., Ruurlo, Netherlands). Before heating, the N2 gas was humidified to 90 % (R.H., 25 °C) using a glass fritted bubbler. After vaporization, the gas was split between the ionization source and an exhaust line by a T-piece. This split was found to be necessary to ensure an adequate vaporization of the eluent phase, without significantly compromising the overall sensitivity of the method: the dilution factors were 4.3 and 2 for the LCQ and LTQ-Orbitrap, respectively. All the concentration values reported in this work are liquid concentrations and are not re-calculated based on the split factors.
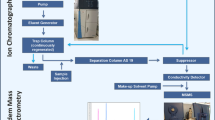
Optimization of the method was carried out on an LCQ Deca XP. A schematic of the sample introduction system is shown in Fig. 1. This system generates a constant gas-phase concentration of molecules. Since the sample flow inside the capillary is regulated by applying a defined pressure of inert gas (N2) into the sample vial, the gas-phase concentration can be easily calculated using Poiseuille’s formula, if the capillary dimensions and the applied pressure are known. In this study, all reported concentrations are liquid concentrations.

(Top) Schematic of the setup employed in this study. The nano-LC capillary column is directly inserted in the vaporization chamber at a defined depth. A concentric flux of humidified and heated nitrogen allows complete vaporization of the liquid phase. The vapors enter the DBDI source and analytes are thereby ionized; (bottom) simplified schematic (not in scale) of the DBD electrodes
The LC system employed was an Eksigent Ekspert nano-LC 400 (AB Sciex, Darmstadt, Germany). The capillary column was a reverse-phase C18 (3 μm) 75 μm × 15 cm column (AB Sciex), which was held at ambient temperature (19–21 °C) during all the analyses. A binary gradient was used: water/acetonitrile 95:5 (solvent A, 0.05 % formic acid) and acetonitrile (solvent B, 0.05 % formic acid). The solvent gradient was chosen to deliver a good separation of the different pesticides, while keeping the total analysis time as short as possible. The LC program was started with 100 % solvent A for the first minute, then ramped linearly from 90 to 30 % A until 18 min, and then kept for 2 min at 30 %. Within 1 min, the initial conditions were then restored. Column equilibration time was 5 min. The total nano-LC run time was 33 min, including sample injection and column equilibration. Sensitivity was improved by performing a large-volume full-loop injection of 5 μL at a flow of 800 nL/min. The effects of the sample solvent composition on the LC peak shape were investigated and are reported in Fig. S1 in the Electronic Supplementary Material (ESM). When a 9:1 H2O/ACN ratio is used as eluent, peaks were narrow and their shape did not depend much on the injection volume.
Two mass spectrometers were used in this study: a Thermo LCQ Deca XP and a Thermo LTQ-Orbitrap (Thermo Fischer Scientific). The total gas flow entering the source was constant and dictated by the inlet of the mass spectrometer, i.e., the metal transfer capillary dimensions. The measured flows were 0.7 L/min for the LTQ-Orbitrap and 1.5 L/min for the LCQ Deca XP. Initial optimization of the ionization source and the vaporization setup was performed with the LCQ Deca XP. The MS interface parameters were as follows: capillary voltage, 8.05 V; tube lens voltage, 40 V; capillary temperature, 275 °C; and lens voltage, −30.67 V. The acquisition was performed with a mass window of 100 to 400 m/z in “profile mode,” with three microscans averaged, with a maximum injection time of 50 ms, and with automated gain control (AGC) on.
Quantification was performed with a Thermo LTQ-Orbitrap in full scan mode, with centroid acquisition and a mass resolution of 30,000 FWHM at 400 m/z. The MS interface parameters were as follows: capillary voltage, 8.05 V; tube lens voltage, 60 V; and capillary temperature, 275 °C. The acquisition was performed with a mass window of 150 to 350 m/z, with one microscan, and with a maximum injection time of 500 ms. AGC was set on.
Depending on the resolving power of a mass spectrometer, one can perform quantitative experiments based on tandem mass spectrometry (for low-resolution instruments) or on full scan spectra (for high-resolution ones). The hybrid LTQ-Orbitrap used is capable of a resolution of up to 100,000 FWHM at 400 m/z. For such mass analyzer, the resolving power affects the required acquisition time [19], i.e., longer acquisition times are needed to obtain more highly resolved spectra. With our version of the Orbitrap, the maximum resolution required an acquisition time of more than 2 s. This time is inadequate to obtain a sufficient number of data points for typical nano-LC peaks. Therefore, the resolution of the LTQ-Orbitrap was lowered to 30,000 resulting in a faster scan speed of 1.4 scans per second. Generally, the mass accuracy achieved was very satisfactory, with a relative error ranging from 0.1 to 0.8 ppm. A mass tolerance of 2 ppm was used for creation of extracted ion chromatograms and signal integration.
Ionization source parameter optimization
The details of the active capillary plasma source are already described elsewhere [17, 18]. Briefly: an AC voltage is applied to two concentric electrodes, separated by a dielectric capillary connected to the MS inlet. The DBDI source mostly generates protonated ions, with very little to no fragmentation (Fig. 2). The plasma characteristics (e.g., density of reactive species and plasma length) depend on the applied voltages, the operating frequency, and the electrode positions. In order to achieve optimal ionization conditions (i.e., highest ion abundance), the electrode positions were carefully optimized (see Figs. S2 and S3 in the ESM). It is clear that ion abundance is maximized for a specific electrode position, which was found to be independent of the analyte. An increase in the applied voltage also led to an increase in signal, which was found to be somewhat compound dependent (ESM Fig. S3). Best results were obtained with an applied voltage of 1.6 kVp-p. The operating frequency was set to 5.75 kHz. We would like to emphasize that this low-temperature plasma ionization source can operate at room temperature with very similar performance. In this work, a temperature of 260 °C was used with the only purpose of ensuring an adequate vaporization of the eluent phase.

Nano-LC-DBDI full scan mass spectra of atrazine and carbofuran, at a concentration of 100 ng/mL
Results and discussion
The reported coupling of a nano-LC system with an ambient DBDI source is novel in several respects. First of all, with few exceptions [14–16], it is rare that an LC system is directly interfaced to a DBDI source. In addition, food analysis is mainly performed by HPLC and UPLC but rarely done by nano-LC. We used the potential of both nano-LC and DBDI to achieve an extraordinary (pg/g) sensitivity for pesticide analysis. This was possible because of the chromatographic separation and enrichment provided by the nano-LC and due to its low solvent flow rate, an important condition to avoid suppression effects during the ionization in the DBD source. In our system, the higher flows delivered by UPLC would have caused extensive ion suppression, compromising the quantitation.
The selected pesticides have a medium to high polarity and belong to different chemical classes, i.e., triazine, carbamate, organophosphate, phenylamide, substituted urea, and chloroacetanilide. It is known that DBDI is particularly sensitive towards mid-polar to polar compounds (especially when containing N, S, or P atoms) and less sensitive for non-polar compounds like organochlorine pesticides, a limitation intrinsic in most DBD plasmas. Nevertheless, this method is very sensitive towards pesticides that usually require derivatization prior to GC (e.g., carbamates) or non-GC-amenable ones, like the neonicotinoid imidacloprid (Fig. 3). In this last case, the LOD achieved with direct liquid injection was 30 ng/mL, which would correspond approximately to 300 pg/mL with a nano-LC chromatographic focusing of the injected liquid.

Positive ion mode MS/MS spectrum of imidacloprid at a concentration of 100 ng/mL, acquired with a Thermo LTQ (collision energy 20, manufacturer’s units). The sample was directly introduced inside the heating chamber through a fused silica capillary connected to the nano-LC. The flow rate was 800 nL/min. The LOD was 30 ppb
A nano-LC-DBDI-MS chromatogram of the analysis of a 100-ng/mL pesticide sample mix is shown as TIC in Fig. 4 together with some extracted ion chromatogram (EIC) traces.

Nano-LC-DBDI-MS chromatogram of a mix of the pesticides listed in Table 1 (top) and extracted ion chromatograms of the quasimolecular ions of some of them (bottom). Injection volume was 5 μL, at a concentration of 100 ng/mL, with a 9:1 H2O/ACN (v/v) solvent composition
Calibration curves were measured with water/acetonitrile 9:1 (v/v) solutions, spiked with a mix of the 16 pesticides at concentration levels from 10 pg/mL to 1 μg/mL. Analyses were performed in triplicate for each concentration level. A 1/x 2 weighting was used in the linear regression. Blank samples were injected during the calibrations to verify that no carry-over was present.
For quantification, EIC peaks were integrated for each pesticide. Only the quasimolecular ions [M+H]+ were used, since negligible fragmentation was observed for all pesticides. The ionization is soft due to the peculiar characteristics of our low-temperature plasma. It is generated between two ring-shaped electrodes, and the residence time of the gas-phase analytes in the plasma itself is very low. This results in a very low energy transfer from the plasma to the molecules, allowing them to ionize but not fragment significantly. Even for very labile compounds, e.g., 2,4,5-trichlorophenoxyacetic acid, the in-source fragmentation is contained and not much more extensive than in ESI (see ESM Fig. S4). In this case, the extent of fragmentation did not depend much on the vaporization temperature, but was an effect of the transit of the molecules inside the reactive plasma. The LOD was 3 μg/mL, which would correspond approximately to 30 ng/mL with a nano-LC chromatographic focusing of the injected liquid.
The behavior of our plasma ionization is very reproducible. Even without internal standardization, the signal linearity was very satisfactory in the concentration range investigated (see linear regression parameters in Table 1). The best sensitivity was observed for metalaxyl, a phenylamide fungicide. In this case, the linear dynamic range was from 1.8 ng/mL to 1 μg/mL, covering almost three orders of magnitude. A very high sensitivity was also observed for the triazine pesticides, i.e., terbuthylazine, atrazine, and simazine (reported in order of decreasing sensitivity). In this last case, a direct comparison of the sensitivity is possible, since those molecules only differ for a substituent on the triazine ring: terbuthylazine is the most basic of those and therefore is more easily protonated inside the reactive plasma. On the other hand, a lower sensitivity was observed for propham and bendiocarb. In this last case, the linear dynamic range was limited up to 300 ng/mL, while for all the other pesticides the signal linearity was up to 1 μg/mL.
The reported sensitivity is in general superior to the one obtained with LC-MS/MS or GC-MS [1] (see Table 1 for a detailed comparison). It also outperforms similar LC-DBDI-based techniques, e.g., the ones reported by Gilbert-López et al. [14, 16]. Regarding the first work [16], a direct comparison of the two ionization sources is possible, since in both cases an Orbitrap mass analyzer was used: the reported LODs in a spiked olive oil extract for simazine, terbuthylazine, and malathion were 500, 500, and 25,000 pg/g, respectively, values that are considerably higher than the ones obtained within this work for the same analytes, i.e., 30, 10, and 100 pg/mL, respectively. The LOQs reported by the same author [14] in an orange juice extract for atrazine (10,000 pg/g), dichlorvos (10,000 pg/g), isoproturon (10,000 pg/g), metalaxyl (19,000 pg/g), simazine (10,000 pg/g), and terbuthylazine (16,800 pg/g) are on average more than 60 times higher than the ones obtained in this work. In the latter case, however, a direct comparison is not possible because a different mass analyzer was used.
For better illustration, two example calibration plots are shown in Fig. 5. Since no background signal was observed in the EICs, limits of detection (LODs) and quantification (LOQs) were calculated from the regression equations, according to the formulae:
where S B represents the standard deviation of the intercept and A the slope of the curve. Limits of detection were also determined experimentally by analyzing samples of decreasing concentration, until no signal was recorded.

Calibration curves for atrazine and carbofuran. Top shows linear-linear and bottom, log-log plots
The LOQs were all easily compliant and way below the current European Union MRLs for apples, reported in Table 1. Such high sensitivity is particularly important when samples should be pesticide free, e.g., organic food, or where their level should be much below the maximum legal limits, e.g., in baby food. It is important to underline that the very low LODs achieved did not result from sample preconcentration using SPE or similar techniques. We also note that the use of triple quadrupole instrumentation, often employed for quantification purposes, could further decrease the reported LODs.
Extraction efficiency, matrix effects, and intra-day reproducibility
QuEChERS extraction is nowadays a reliable and widespread technique employed for pesticide analysis. The extraction efficiency was calculated for a concentration of 10 ng/mL for all the 16 pesticides. Values ranged from 82 to 108 % (ESM Fig. S5).
Matrix effects were evaluated for organic apples, extracted with the QuEChERS procedure mentioned above. After dilution of the sample extract with water, using the same 9:1 water/acetonitrile ratio as for the calibration, it was spiked at a final concentration of 10 ng/mL. Therefore, the dilution factor of the matrix extract was equal to 10. The matrix-matched sample spikes were injected in triplicate, and the instrumental response was compared to the one from the injected spiked pure solvent samples. Matrix effects were calculated as follows:
were spikein matrix and spikein solvent represent the peak areas for the spiked pesticide in pesticide-free matrix and in pure solvent, respectively. The results are reported in Fig. 6. No significant suppression/enhancement effect was observed for most of the pesticides, with less than ±5 % for nine of them. An enhancement effect of 9–10 % was observed for ethiofencarb and phoxim. Bendiocarb was suppressed by 15 % according to our measurements. A stronger enhancement effect of 21 and 29 % was observed for isoproturon and malathion, respectively. The average matrix effect is calculated to be ±7.7 % for all pesticides investigated. This is quite satisfactory, considering that no sample cleanup (dispersive-SPE step) was carried out. Further improvement and exclusion of matrix effects could be achieved with labelled internal standardization and by performing matrix-matched calibration. Nevertheless, our results confirm the generally very high robustness of the DBDI-nano-LC coupling and its applicability to real world samples. As an example, due to the very high sensitivity, one could easily perform direct analysis of water samples without previous up-concentration by SPE: water samples can be directly injected into the nano-LC column after filtration with a 0.2-μm Teflon filter, and in this case, even higher sample volumes can be used for on-column focusing, because of the absence of organic solvents in the sample.

Matrix effect measured for the different pesticides spiked at a concentration level of 10 ng/mL (n = 3)
Intra-day reproducibility was calculated by analyzing blank apple extracts, spiked with the mix of pesticides considered to a final concentration of 10, 1, and 0.1 ng/mL. The blank acetonitrile extracts were diluted with chromatography water in the same manner as for the matrix effect calculation, in order to have a final solvent composition of 9:1 H2O/ACN (v/v). Nano-LC analyses were performed in triplicate and with the same experimental conditions used for the calibration curve.
It is important to note that even in the case of the matrix effect, extraction efficiency and reproducibility experiments, for most of the analytes, the EIC traces did not show background noise in their elution proximities. This could be explained with an efficient separation of pesticides from matrix components, the lower volatility of matrix interferences compared to the pesticides, as well as the use of the centroid MS acquisition.
Conclusions
We report the first coupling of a nano-LC system to an ambient ionization source. To the best of our knowledge, this is the most sensitive and reproducible DBDI-MS-based method for pesticide analysis reported to date. It matches or outperforms the sensitivity of current LC-ESI-MS/MS and GC-EI-MS methods [1] in the analysis of mid-polar to polar compounds, and could therefore be proposed as an alternative method for routine pesticide analysis. One additional advantage of our system is the possibility to detect compounds not only in liquids but also in gas-phase samples, giving more flexibility in the choice of the analytical method.
In our work, the use of liquid chromatography gave the following advantages: first, the sensitivity was much higher compared to a direct infusion of a liquid sample, where pesticides can be normally detected only at low nanogram-per-milliliter concentration levels using our setup. Another advantage is due to the chromatographic step itself, which helps in the identification of the compounds based on retention time, especially when high-resolution MS instrumentation is not available and tandem mass spectrometry has to be used. The most important advantage was the reduction of ion suppression, which in our system was pronounced when different pesticides at high concentration levels (above 1 μg/mL) were simultaneously ionized. In fact, during the ionization process, analytes (as well as matrix components) compete for the available protons in the reactive plasma. A complete or partial chromatographic separation prior to ionization allowed reducing these suppression phenomena to a negligible level.
We found a large linear dynamic range, covering almost four orders of magnitude for most analytes. In addition, very low limits of detection, as low as 10 pg/mL and 50 fg on-column were achieved. The low matrix effect experienced upon analysis of real samples also suggests this method for more complex matrices.
The average repeatability of 3.9, 17.6, and 21.2 % RSD at 10, 1, and 0.1 ng/mL, respectively, is remarkable, considering the use of an ambient ionization technique. Herein also lies the “magic” of the high sensitivity, since the active capillary plasma ionization features an “in line” geometry, which causes the ions to be formed within the inlet capillary; the transmission into the MS and thereby the sensitivity are greatly enhanced (virtually 100 %).
In this report, we demonstrated a robust and very sensitive analytical method based on an ambient ionization nano-LC coupling and its application for different classes of pesticides, including triazines, carbamates, and organophosphorus pesticides, which could be easily employed by conventional laboratories already using the standard QuEChERS extraction procedure.
References
Alder L, Greulich K, Kempe G, Vieth B. Residue analysis of 500 high priority pesticides: better by GC-MS or LC-MS/MS? Mass Spectrom Rev. 2006;25(6):838–65.
Karlsson KE, Novotny M. Separation efficiency of slurry-packed liquid chromatography microcolumns with very small inner diameters. Anal Chem. 1988;60(17):1662–5.
Hernández-Borges J, Aturki Z, Rocco A, Fanali S. Recent applications in nanoliquid chromatography. J Sep Sci. 2007;30(11):1589–610.
Mills M, Maltas J, Lough WJ. Assessment of injection volume limits when using on-column focusing with microbore liquid chromatography. J Chromatogr A. 1997;759(1):1–11.
Buonasera K, D’Orazio G, Fanali S, Dugo P, Mondello L. Separation of organophosphorus pesticides by using nano-liquid chromatography. J Chromatogr A. 2009;1216(18):3970–6.
Cappiello A, Famiglini G, Palma P, Mangani F. Trace level determination of organophosphorus pesticides in water with the new direct-electron ionization LC/MS interface. Anal Chem. 2002;74(14):3547–54.
Cappiello A, Famiglini G, Mangani F, Palma P, Siviero A. Nano-high-performance liquid chromatography–electron ionization mass spectrometry approach for environmental analysis. Anal Chim Acta. 2003;493(2):125–36.
Zhu KY, Leung KW, Ting AKL, Wong ZCF, Ng WYY, Choi RCY, et al. Microfluidic chip based nano liquid chromatography coupled to tandem mass spectrometry for the determination of abused drugs and metabolites in human hair. Anal Bioanal Chem. 2012;402(9):2805–15.
Asensio-Ramos M, D’Orazio G, Hernandez-Borges J, Rocco A, Fanali S. Multi-walled carbon nanotubes-dispersive solid-phase extraction combined with nano-liquid chromatography for the analysis of pesticides in water samples. Anal Bioanal Chem. 2011;400(4):1113–23.
Smoluch M, Mielczarek P, Silberring J. Plasma‐based ambient ionization mass spectrometry in bioanalytical sciences. Mass Spectrom Rev. 2015. doi:10.1002/mas.21460.
Wiley JS, García-Reyes JF, Harper JD, Charipar NA, Ouyang Z, Cooks RG. Screening of agrochemicals in foodstuffs using low-temperature plasma (LTP) ambient ionization mass spectrometry. Analyst. 2010;135(5):971–9.
Albert A, Kramer A, Scheeren S, Engelhard C. Rapid and quantitative analysis of pesticides in fruits by QuEChERS pretreatment and low-temperature plasma desorption/ionization orbitrap mass spectrometry. Anal Methods. 2014;6(15):5463–71.
Tang F, Chen J, Wang X, Zhang S, Zhang X. Development of dielectric-barrier-discharge ionization. Anal Bioanal Chem. 2015;407(9):2345–64.
Gilbert-López B, Geltenpoth H, Meyer C, Michels A, Hayen H, Molina-Díaz A, et al. Performance of dielectric barrier discharge ionization mass spectrometry for pesticide testing: a comparison with atmospheric pressure chemical ionization and electrospray ionization. Rapid Commun Mass Spectrom. 2013;27(3):419–29.
Hayen H, Michels A, Franzke J. Dielectric barrier discharge ionization for liquid chromatography/mass spectrometry. Anal Chem. 2009;81(24):10239–45.
Gilbert-López B, García-Reyes JF, Meyer C, Michels A, Franzke J, Molina-Díaz A, et al. Simultaneous testing of multiclass organic contaminants in food and environment by liquid chromatography/dielectric barrier discharge ionization-mass spectrometry. Analyst. 2012;137(22):5403–10.
Nudnova MM, Zhu L, Zenobi R. Active capillary plasma source for ambient mass spectrometry. Rapid Commun Mass Spectrom. 2012;26(12):1447–52.
Wolf J, Schaer M, Siegenthaler P, Zenobi R. Direct quantification of chemical warfare agents and related compounds at low ppt levels: comparing active capillary dielectric barrier discharge plasma ionization and secondary electrospray ionization mass spectrometry. Anal Chem. 2014;87(1):723–9.
Makarov A, Scigelova M. Coupling liquid chromatography to Orbitrap mass spectrometry. J Chromatogr A. 2010;1217(25):3938–45.
Acknowledgments
The authors acknowledge Dr. Juan Zhang from Novartis AG for the generous donation of the high-resolution Orbitrap-MS system used within this study. They also thank Christian Marro from the technical workshop of ETH for the valuable support and the construction of the experimental setup.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 647 kb)
Rights and permissions
About this article

Cite this article
Mirabelli, M.F., Wolf, JC. & Zenobi, R. Pesticide analysis at ppt concentration levels: coupling nano-liquid chromatography with dielectric barrier discharge ionization-mass spectrometry. Anal Bioanal Chem 408, 3425–3434 (2016). https://doi.org/10.1007/s00216-016-9419-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9419-x