
Abstract
A freeze-dried mussel tissue (Mytilus edulis) reference material (CRM-FDMT1) was produced containing multiple groups of shellfish toxins. Homogeneity and stability testing showed the material to be fit for purpose. The next phase of work was to assign certified values and uncertainties to 10 analytes from six different toxin groups. Efforts involved optimizing extraction procedures for the various toxin groups and performing measurements using liquid chromatography-based analytical methods. A key aspect of the work was compensating for matrix effects associated with liquid chromatography-mass spectrometry through standard addition, dilution, or matrix-matched calibration. Certified mass fraction values are reported as mg/kg of CRM-FDMT1 powder as bottled for azaspiracid-1, -2, and -3 (4.10 ± 0.40; 1.13± 0.10; 0.96 ± 0.10, respectively), okadaic acid, dinophysistoxin-1 and -2 (1.59 ± 0.18; 0.68 ± 0.07; 3.57± 0.33, respectively), yessotoxin (2.49 ± 0.28), pectenotoxin-2 (0.66 ± 0.06), 13-desmethylspirolide-C (2.70 ± 0.26), and domoic acid (126 ± 10). Combined uncertainties for the certified values include contributions from homogeneity, stability, and characterization experiments. The commutability of CRM-FDMT1 was assessed by examining the extractability and matrix effects for the freeze-dried material in comparison with its equivalent wet tissue homogenate. CRM-FDMT1 is the first shellfish matrix CRM with certified values for yessotoxins, pectenotoxins or spirolides, and is the first CRM certified for multiple toxin groups. CRM-FDMT1 is a valuable tool for quality assurance of phycotoxin monitoring programs and for analytical method development and validation.

CRM-FDMT1 is a multi-toxin mussel tissue certified reference material (CRM) to aid in development and validation of analytical methods for measuring the levels of algal toxins in seafood
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liquid chromatography-mass spectrometry (LC-MS) has proven to be a powerful technique for the analysis of multiple phycotoxins in a single run and has been implemented in many shellfish monitoring laboratories worldwide [1–5]. Recently, LC-MS has replaced the rodent bioassay as the official reference method for monitoring lipophilic algal toxins in shellfish [6]. Certified reference materials (CRMs) containing these toxins in shellfish tissue matrices are required for development, validation, and quality assurance of extraction and detection methods [7]. Some matrix CRMs are available for individual toxin groups such as domoic acid, okadaic acid, and azaspiracids [8–10]. While these CRMs provide the required traceability for their certified analytes, matrix materials for other regulated toxins (e.g., yessotoxins and pectenotoxins) remain unavailable. With the increased use of LC-MS for multi-toxin monitoring, the need for a CRM certified for multiple groups of toxins has also increased. An international collaboration to produce a freeze-dried mussel tissue (Mytilus edulis) CRM (CRM-FDMT1) [11] was initiated following a series of feasibility studies [7, 11–15]. This material contains notable levels of analytes from six major groups of shellfish toxins, including domoic acid (DA), azaspiracids (AZAs), okadaic acid (OA) and dinophysistoxins (DTXs), pectenotoxins (PTXs), yessotoxins (YTXs), and spirolides (SPXs). Development of extraction and LC-MS methods [16] preceded a comprehensive assessment of the homogeneity and stability of CRM-FDMT1 [17]. Homogeneity was found to be excellent across the entire production series, while stability studies showed no degradation at temperatures as high as +18 °C over 1 y.
This paper describes the efforts in assigning certified mass fraction values and associated uncertainties to a series of toxins present in CRM-FDMT1 (Table 1). For certification purposes the toxins were measured using a group-wise approach, employing optimized LC-MS/MS methods and fully exhaustive extraction procedures. A variety of measures that included standard addition, matrix-matched calibration, and dilution were taken to compensate for matrix effects associated with LC-MS [16]. Combined uncertainties were assigned to include contributions from characterization, homogeneity, and stability. An assessment of the commutability of CRM-FDMT1 was made in terms of toxin extractability and instrumental response characteristics.
Materials and methods
Standards and chemicals
Methanol, acetonitrile, and trifluoroacetic acid (TFA) were purchased from Caledon (Georgetown, ON, Canada). Ammonium acetate, sodium hydroxide, hydrochloric acid, and formic acid were purchased from Fluka (Oakville, ON, Canada). Ammonium formate (97 %) was purchased from Sigma-Aldrich (Oakville, ON, Canada). Bakerbond Octyl (C8) 40 μm prep LC packing was purchased from Krackeler Scientific, Inc. (Albany, NY, USA). Shellfish toxin CRMs were obtained from the National Research Council (NRC, Halifax, NS, Canada; www.nrc.ca/crm). All water used in these experiments was from a Milli-Q gradient A10 purification system (Millipore Corp., Billerica, MA, USA).
Extraction procedures
CRM-FDMT1 is provided in bottles with a sufficient quantity of powder for multiple sub-samples. Based on the results of a sub-sampling study, a minimum sub-sample size of 0.35 g is recommended (reconstituted to 2 g with water) [16]. To ensure representative sub-sampling of CRM-FDMT1, bottles were brought to room temperature and the contents mixed thoroughly by rolling and inversion before opening. The moisture content of the material has been previously assessed [11], and certified values are based on the mass of the freeze-dried powder as provided in the bottles. Portions (0.35 g or 0.7 g) were measured into 50 mL centrifuge tubes, recording weights on an analytical balance, before reconstitution by adding water (1.65 mL or 3.3 mL, respectively) and vortex mixing for 30 s followed by sonication for 60 s. Specific details of extraction conditions for the different toxin groups are provided (Table 2), while the general procedures are described below. Existing matrix CRMs for some of the individual toxin groups (CRM-ASP-Mus-d; CRM-DSP-Mus-b; CRM-AZA-Mus) were also prepared as part of these experiments. All extracts were passed through 0.45 μm regenerated cellulose syringe filters (Millipore Corp, Billerica, MA, USA) and stored at –20 °C until analysis.
Liquid-solid extraction (LSE)
The appropriate extraction solvent (Table 2) was added to reconstituted CRM-FDMT1 in 50 mL centrifuge tubes (2 g for lipophilic toxins, 4 g for DA), vortexed at 2500 rpm for 3 min (model DVX-2500; VWR Int., Radnor, PA, USA), then centrifuged at 3950 × g for 10 min (Sorvall Legend RT+; Thermo Scientific, Osterode, Germany). The supernatant was transferred to a volumetric flask (25 mL for lipophilic toxins, 50 mL for DA) and the remaining CRM-FDMT1 pellet subjected to three further extractions steps. Supernatants were combined and made to volume in the volumetric flask. Toxin recovery was assessed by extracting the remaining pellet with a final 4.5 mL of solvent, making to 5 mL volume in a separate volumetric flask and measuring for the presence of residual analyte.
Matrix solid-phase dispersion (MSPD)
MSPD procedures were based on methods developed for shellfish toxins [10]. An aliquot (0.5 g) of reconstituted CRM-FDMT1 was weighed together with 2 g of the Bakerbond C8 stationary phase (Table 2) in a polystyrene weighing boat. This was then transferred to a glass mortar and ground with a pestle until homogenous. The mix was loaded into a 7 mL glass solid phase extraction (SPE) column prepacked with 0.2 g of clean stationary phase placed between two frits. Packed tubes were then placed on vacuum manifold for elution with 10 mL of the appropriate extraction solvent (Table 2) into a volumetric flask. To assess the method for quantitative recovery, an additional 5 mL of extraction solvent was passed through the extracted SPE column, made to 5 mL volume in a volumetric flask and an aliquot filtered before measuring for the presence of residual analyte.
Instrumental analyses
LC-MS analyses were conducted on an Agilent 1200 LC system (Agilent Inc., Palo Alto, CA, USA), equipped with a binary pump, degasser, column oven, and autosampler, connected to an API4000 QTRAP mass spectrometer equipped with a Turbospray ionization source (Sciex, Concord, ON, Canada). LC-UV analyses were conducted on an Agilent 1200 LC system was equipped with a model G1315D diode array detector (set at 242 nm).
LC separations of lipophilic toxins were achieved using gradient elution with either neutral or acidic mobile phases. Isocratic elution with an acidic mobile phase was used for DA and epi-DA. Toxins were analyzed by MS using selected reaction monitoring (SRM) with optimized collision energy (CE) and declustering potential (DP) settings. Details of instrumental conditions for each analyte group are shown in Table 2.
Compensation for matrix effects in LC-MS
Standard addition
Extracts were spiked (20 % v/v) using a Hamilton Microlab to dispense precise volumes. CRM-FDMT1 extracts were spiked with either methanol or CRM stock solutions prepared gravimetrically in methanol ensuring the same matrix concentration in the samples. The CRM spike levels were prepared such that the spiked concentrations were approximately double the concentration of methanol spiked extracts. Linearity of response for toxins in the CRM-FDMT1 matrix by LC-MS was demonstrated previously [16], so a single spike level was used in these standard addition experiments. Triplicate spikes of both methanol and CRM stocks were made for each sample extract, with three to four sample extracts prepared by each extraction method.
Matrix-matched calibration
Stock solutions were prepared gravimetrically from calibration solution CRMs, using methanol as diluent. The stock solution was then serially diluted 3-fold with methanol using the Hamilton Microlab. A six-level series was spiked into CRM-Zero-Mus extracts (20 % v/v) to create matrix-matched calibrants with concentrations ranging from near the method’s limit of quantitation to approximately twice the toxin concentration in CRM-FDMT1 extracts (MSPD or LSE), which were diluted by 20 % (v/v) with methanol to maintain equivalent matrix concentrations to the calibrants.
Dilution
CRM-FDMT1 extracts were diluted with methanol using the Hamilton Microlab. The range of dilution depended on the MS response available for each analyte.
For all preparations using the Hamilton Microlab, the amount collected and dispensed was recorded gravimetrically to determine volumes accurately.
Base hydrolysis
A base hydrolysis procedure was adapted from previous work [18]. Aliquots (1 mL) of LSE extracts (n = 3) were hydrolyzed with 125 μL 2.5 M NaOH for 40 min at 70 °C before neutralizing with 125 μL 2.5 M HCl. The samples were filtered and analyzed using the conditions outlined in Table 2 for OA/DTXs.
Commutability
CRM-FDMT1 was compared to samples of the wet tissue homogenate used to produce the CRM [11]. Samples of this wet material had been stored at –20 °C. DA was chosen as a test analyte. Samples from five bottles of CRM-FDMT1 and five sub-samples of the wet homogenate were extracted and measured by LC-UV and LC-MS. The equivalence of instrumental response for lipophilic analytes by LC-MS was compared by performing standard additions on extracts of CRM-FDMT1, the initial wet tissue homogenate, and CRM-Zero-Mus.
Results and discussion
Certification measurements and uncertainty calculations
Measurements for assigning certified values to reference materials must be made in such a way that final property values are traceable to an accurate realization of the unit in which property values are expressed [19]. To ensure confidence in assigned values, it is ideal if two or more independent methods are used to generate results from which the property value is assigned. With this approach, each method would use independent sources of calibrants and would ideally be verified with traceable quality control samples (i.e., separate CRMs).
An approach was taken in which key steps of the analytical process were varied in order to have independent analytical approaches for assignment of the certified values to CRM-FDMT1. For extraction, LSE procedures are commonly used for shellfish toxin determination [2, 5, 20, 21], and recent work has shown the utility of MSPD in the analysis of these compounds as well [10]. LC-MS is the only detection and measurement technique offering sufficient sensitivity and precision for most of the target analytes in CRM-FDMT1. Although a multi-toxin LC-MS method was developed earlier in this project [16] for use in homogeneity and stability testing [17], the extraction and analysis methods needed to be further refined for specific toxin groups in order to achieve the level of extraction efficiency favorable for certification. Toxin-specific LSE and MSPD procedures were optimized to exhaustively extract individual toxin groups in CRM-FDMT1. Vortex mixing versus shear homogenization, the number of extraction steps, and solvent composition were evaluated for the LSE method. The elution volume and solvent composition were tested for the MSPD method. Extraction efficiency was verified through analysis of analytes in the additional extraction aliquots.
The accuracy of calibrating with solution CRMs to determine property values of analytes in natural extracts is an important consideration, particularly for determinations by MS with electrospray ionization where matrix effects can cause a significant bias [22, 23]. Approaches such as standard addition and matrix-matched calibration were selected to compensate for matrix effects during certification measurements. It was possible to eliminate matrix effects for some analyte groups by diluting extracts and using external calibration with solvent standards [see Electronic Supplementary Material (ESM)]. Table 2 summarizes the selected analytical approaches used to assign certified values to the respective toxin groups present in CRM-FDMT1.
Property values and associated uncertainties were assigned in accordance with internationally recognized procedures and guidelines. Raw data were rigorously evaluated by outlier testing [24] and property values were assigned by taking the unweighted mean of the two methods’ final results. Calculation of the overall expanded uncertainty (U CRM) for each analyte (Eq. 4.1) involved applying a coverage factor (k = 2) to the standard uncertainty (u CRM):
where u CRM considers uncertainty contributions from characterization (u char), homogeneity (u hom), and stability (u lts & u sts) by applying the relative uncertainties from these experiments to the newly assigned property value:
Various sources of uncertainty relating to calibration were considered, including the uncertainty associated with the calibration solution CRMs used, the uncertainty associated with calibrant preparations (i.e., dilutions, spiking), as well as uncertainties from measurements of the calibrant itself. A detailed description of the uncertainty approach applied for CRM-FDMT1 is provided in the ESM.
Azaspiracids
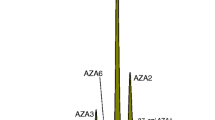
CRM-Zero-Mus was shown to be an appropriate matrix for preparation of matrix-matched calibrants for AZAs in CRM-FDMT1 [16]. This work also demonstrated that extracts spiked with AZA calibrant CRMs are linear at up to three times the CRM-FDMT1 concentrations in LSE extracts. LSE was used with standard addition and MSPD with matrix-matched calibration to assign values. Good agreement between data obtained from the two different approaches (Table 2) provided confidence in the accuracy of the certified values. The experiments were verified through parallel analysis of CRM-AZA-Mus [10] using the same procedures. Other experiments, including LSE with matrix matched calibration, MSPD with standard addition, and both LSE and MSPD with dilution, were conducted to support the values used for certification (ESM). The neutral pH mobile phase provided resolution of AZA epimers [25] from the parent peak as established previously [16] (Fig. 1A). Molar responses of the 37-epi-AZAs [25] are approximately equivalent to the parent toxins under the LC-MS conditions used. Epimer peak areas corresponded to approximately 6.5–7.5 % of total peak areas for AZA1, -2, and -3. For certification purposes, the epimer and parent toxin peaks were integrated and combined to assign the certified values. Certified concentrations are shown with associated combined uncertainties in Table 3.

LC-MS chromatograms for lipophilic toxin groups in CRM-FDMT1 (conditions: Table 2). Epimer peaks for AZA-1, -2, and -3 are combined with the main peaks to assign certified values. AZA6 is not certified and later eluting peaks in AZA3 and AZA6 traces (*) are degradation products from thermal sterilization of CRM-FDMT1 (A). OA and DTX2 isomer peaks (*) are not included in certified value (B). YTX and PTX2 analysis in successive negative and positive ionization periods. PTX2 isomers (*) are not part of the certified value. Peak at 6.1 min increases after exposure to acidic conditions suggesting it is a spiroketal isomer of PTX2 [30] (C). LC-MS/MS analysis of 13-desMe-SPX-C (D)
Okadaic acid and dinophysistoxins
OA, DTX1, and DTX2 were quantitated by LC-MS (Fig. 1B) using standard addition for both LSE and MSPD extracts. In developmental work, it was found that matrix-matched calibration with CRM-Zero-Mus was applicable for OA but less suitable for DTX2 and DTX1. Given this, OA quantitation by matrix-matched calibration provided supporting data in excellent agreement with values obtained by standard addition (ESM), with reduced agreement for DTX2 and DTX1 given the slight differences in matrix effects. Dilution was assessed as a method for eliminating matrix effects for OA group toxins and was found to be unsuitable. The extent of dilution required to completely eliminate matrix effects was not feasible considering the MS signal available under the described conditions. Concentrations measured for the OA group toxins in CRM-FDMT1 are shown in Table 2 for each method. Assigned certified values and uncertainties are shown in Table 3.
Yessotoxin and pectenotoxin-2
Improved recovery of YTXs was achieved with a slightly aqueous extraction medium without significantly impacting PTX2 recoveries. LSE and MSPD procedures were used for YTX and PTX2 with 80 % and 90 % methanol as extraction solvents, respectively (Table 2), Given both analytes could be adequately extracted with the same procedures and were easily separated chromatographically [16], they were analyzed together. Standard addition was carried out for both analytes on LSE and MSPD extracts. Matrix-matched calibration with CRM-Zero-Mus was not suitable for either YTX or PTX2, while the analyte concentrations were not sufficient for complete elimination of matrix effects through dilution. There was good agreement for YTX and PTX2 concentrations measured in CRM-FDMT1 for both LSE and MSPD extraction methods (Table 2). Assigned certified values and uncertainties are shown in Table 3.
13-Desmethylspirolide-C
13-desMe-SPX-C elicited a strong response in positive mode electrospray ionization and with the relatively high level in CRM-FDMT1, it was possible to eliminate matrix effects entirely through dilution. Matrix effects were negligible beyond a 10-fold dilution (ESM). Extracts were diluted 15-fold and quantitated by external single point calibration. Different calibration solutions were used for LSE and MSPD extracts, prepared at concentration levels near those of the diluted extracts. Results were in excellent agreement for LSE and MSPD extraction methods (Table 2) and the unweighted mean used to assign a final certified value (Table 3). Standard addition was not used for quantitation because previous work suggested a nonlinear response for 13-desMe-SPX-C at higher concentration ranges [16]. In addition, it was found that CRM-Zero-Mus was not a suitable matrix match for spirolide analysis in CRM-FDMT1.
Domoic and C5’epi-domoic acid
The presence of a strong chromophore in DA facilitated quantitative analysis by LC-UV [26, 27]. LC-UV and LC-MS were used as complimentary methods for certification measurements. CRM-FDMT1 samples were prepared using an exhaustive LSE procedure modified from methods developed in the production of an early CRM for DA [8]. LC-MS and LC-UV runs (Fig. 2) were calibrated using external standards prepared from a calibration solution CRM for DA. No significant matrix effects were observed in LC-MS analyses under the conditions used. Results for the two methods used were in excellent agreement (Table 2) and the certified value was assigned as the sum of DA and epi-DA from the unweighted mean results (Table 3).

LC-UV (A) and LC-MS (B) analysis of DA in CRM-FDMT1 (conditions: Table 2). DA isomers in LC-MS trace indicated with an asterisk (*)
Noncertified information values
CRM-FDMT1 was prepared from a variety of materials, including naturally incurred mussel tissues (Mytilus edulis), cultured phytoplankton biomass, and purified or semi-purified toxins [11]. In addition to the certified analytes focused on in this study, CRM-FDMT1 also contains a range of other toxin analogues from each of the different toxin groups represented, as has been highlighted in previous work for AZAs and SPXs in particular [16].
Analysis of the OA group toxins after base hydrolysis revealed the presence of significant quantities of 7-O-acyl esters, particularly for OA and DTX2. Accurate quantitation of these acyl esters remains a significant challenge because of the wide range of ester variants, lack of calibration standards for specific esters, and difficulty in ensuring high-levels of accuracy following the base hydrolysis procedure [18]. Therefore, noncertified information values were assigned to levels of the OA group acyl-esters measured following the base hydrolysis procedure (Table 4).
45-Hydroxy-YTX is present in CRM-FDMT1 and was measured using the same conditions as for YTX (Table 2) with SRM m/z 1157.6 → 1077.6. It was not possible to assign a certified value because a calibration solution CRM is not available for this analyte. An information value of 0.56 mg/kg was measured based on comparison of peak areas with YTX in CRM-FDMT1, assuming equivalent extractability and equimolar MS response.
Commutability
Commutability describes the similarity or equivalence of analytical response obtained for reference materials to the response obtained from routine samples [28, 29]. Freeze-drying and associated processes in the production of CRM-FDMT1 resulted in a significant transformation of the matrix. This concession was considered to be acceptable in order to ensure stability of the toxins over the CRM’s lifetime.
Commutability for CRM-FDMT1 was determined through assessment of DA in the reconstituted CRM matrix and in portions of the initial wet tissue homogenate sampled before freeze-drying. Sub-samples taken at each step of the process indicated greater extraction recovery for CRM-FDMT1 in the first step of the process (Fig. 3). Better recoveries for an extensively processed freeze-dried material are reasonable considering that the small particle size of the powder provided greater surface area to facilitate extraction. However, it is important to note that although recovery was not complete after the first step for either the initial wet tissue or CRM-FDMT1, the overall combined recoveries of the entire extraction process were equivalent for both materials (>99 % efficiency) (Fig. 3). The ratio of the mean result for five replicates of CRM-FDMT1 measured by LC-UV over the mean result for the same replicates measured by LC-MS was 0.99. The ratio for the wet tissue samples was 1.01. The mean results for the separate methods and the variances about these means were considered using a t-test and an F-test, respectively. For both wet and freeze-dried matrices, the F-statistic revealed a significant difference in variances obtained with the two different measurement methods, which is to be expected given LC-UV is inherently more precise for DA analysis. The mean results for LC-UV versus LC-MS were then compared using a t-test assuming unequal variance for both materials. The t-statistics showed that there was no significant difference between the mean results obtained using the two methods for both materials. Given both materials produce similar trends of variance when measured by LC-MS and LC-UV, and the fact that the mean results obtained by LC-UV and LC-MS for both materials were not significantly different from one another, it was concluded that CRM-FDMT1 is commutable with a wet tissue equivalent.

Comparison of DA extractability from CRM-FDMT1 and a wet tissue homogenate. Shown is the % of total extracted from each extraction step. Error bars represent standard deviation from replicate injections of check sample (n = 13)
Further evidence of the equivalence of CRM-FDMT1 to a similar wet tissue homogenate was obtained from studies of matrix effects. Matrix effects were highly comparable between CRM-FDMT1 and its wet precursor material, indicating that production processes did not impact this aspect of the CRM’s commutability.
Traceability
The certified values in CRM-FDMT1 are traceable to the SI system through the use of NRC’s certified calibration standards, and by the use of validated measurement procedures. At all stages in the quantitative analyses, calibrated balances were used to measure sample portions, calibration solution aliquots, and dilutions. Exhaustive extraction procedures were optimized for maximum recovery of analytes. Method accuracy was verified through the use of other shellfish tissue matrix CRMs when available, and by spike-recovery testing where possible. As an example, the assigned AZA values were obtained using two independent calibration methods: matrix-matching using MSPD extracts of CRM-Zero-Mus spiked with NRC certified calibrants; and standard addition by direct spiking of the CRM-FDMT1 extract with NRC certified calibrants. A verification of method accuracy for the spiking process was performed by spiking into clean extraction solvent and quantitating the levels with an external calibration curve. The concentrations of the NRC certified calibrants used were assigned using a quantitative NMR method that was traceable to the SI system through standards produced by National Metrology Institutes that are certified for purity (e.g., NIST SRM-350, benzoic acid). The performance and accuracy of the measurement procedures were also tested using available matrix CRMs such as CRM-AZA-Mus [10], CRM-DSP-Mus-b [9], and CRM-ASP-Mus-d.
Conclusions
This paper represents the culmination of a significant international effort to develop a next generation CRM for shellfish toxins. This is the first shellfish tissue matrix CRM certified for analytes from multiple lipophilic toxin groups, and the first matrix CRM available for yessotoxins, pectenotoxins, and spirolides. Since the levels of many biotoxins in shellfish for human consumption are regulated, CRM-FDMT1 is well suited for use in method development and quality assurance for monitoring programs, particularly where multi-analyte methods such as LC-MS are being used.
CRM-FDMT1 has been certified for AZA1, -2, -3, OA, DTX1, -2, YTX, PTX2, 13-desMe-SPX-C, and DA (mg/kg of powder as bottled). In order to provide the highest quality value assignments, data were generated by two complementary analytical approaches and were supported by additional data from a variety of other experiments (i.e., homogeneity, stability, extraction, and additional certification experiments). Uncertainties were estimated following ISO guidelines and are typical of other shellfish toxin matrix CRMs, with relative expanded uncertainty values ranging from ~6 to 11 % of the certified concentrations.
The levels present in CRM-FDMT1 are relevant to the regulatory levels set forth by agencies such as European Commission and Health Canada. A certified value for 13-desMe-SPX-C should facilitate development and validation of methods for ‘emerging toxins’ such as the cyclic imines. The certified values, the established commutability, and the presence of additional noncertified toxins in CRM-FDMT1 make it a valuable tool for the international analytical science community involved in shellfish toxin determinations.
References
McNabb P, Selwood AI, Holland PT, Aasen J, Aune T, Eaglesham G, et al. Multiresidue method for determination of algal toxins in shellfish: single-laboratory validation and interlaboratory study. J AOAC Int. 2005;88:761–72.
McCarron P, Wright E, Quilliam MA. Liquid chromatography/mass spectrometry of domoic Acid and lipophilic shellfish toxins with selected reaction monitoring and optional confirmation by library searching of product ion spectra. J AOAC Int. 2014;97:316–24.
Quilliam MA, Hess P, Dell'Aversano C (2001) Recent developments in the analysis of phycotoxins by liquid chromatography - mass spectrometry. De Koe WJ, Samson RA, van Egmond HP, Gilbert J, Sabino M (Eds) Proceedings of the 10th International IUPAC Symposium on Mycotoxins and Phycotoxins 21–25 May, 2000 Guaruja (Brazil), pp. 383–391
Gerssen A, Mulder PPJ, McElhinney MA, de Boer J. Liquid chromatography–tandem mass spectrometry method for the detection of marine lipophilic toxins under alkaline conditions. J Chromatogr A. 2009;1216:142–1430.
van den Top HJ, Gerssen A, McCarron P, van Egmond HP. Quantitative determination of marine lipophilic toxins in mussels, oysters and cockles using liquid chromatography-mass spectrometry: inter-laboratory validation study. Food Addit Contam: Part A. 2011;28:1745–57.
Anonymous (2011) Commission Regulation (EU) No. 15/2011 of 10 January 2011 amending Regulation (EC) No. 2074/2005 as regards recognized testing methods for detecting marine biotoxins in live bivalve molluscs. Off J Eur Union L 006 of 11 January 2011:3–6
Hess P, McCarron P, Quilliam MA. Fit-for-purpose shellfish reference materials for internal and external quality control in the analysis of phycotoxins. Anal Bioanal Chem. 2007;387:2463–74.
Hardstaff WR, Jamieson WD, Milley JE, Quilliam MA, Sim PG. Reference materials for domoic acid, a marine neurotoxin. Fresenius J Anal Chem. 1990;338:520–5.
McCarron P, Reeves KL, Giddings SD, Beach DG, Quilliam MA. Development of certified reference materials for diarrhetic shellfish poisoning toxins, Part 2: shellfish matrix materials. J AOAC Int. 2016;99:1163–72.
McCarron P, Giddings SD, Reeves KL, Hess P, Quilliam MA. A mussel (Mytilus edulis) tissue certified reference material for the marine biotoxins azaspiracids. Anal Bioanal Chem. 2015;407:2985–96.
McCarron P, Emteborg H, Nulty C, Rundberget T, Loader JI, Teipel K, et al. A mussel tissue certified reference material for multiple phycotoxins. Part 1: design and preparation. Anal Bioanal Chem. 2011;400:821–33.
McCarron P, Burrell S, Hess P. Effect of addition of antibiotics and an antioxidant on the stability of tissue reference materials for domoic acid, the amnesic shellfish poison. Anal Bioanal Chem. 2007;387:2495–502.
McCarron P, Emteborg H, Hess P. Freeze-drying for the stabilisation of shellfish toxins in mussel tissue (Mytilus edulis) reference materials. Anal Bioanal Chem. 2007;387:2475–86.
McCarron P, Kotterman M, de Boer J, Rehmann N, Hess P. Feasibility of gamma irradiation as a stabilisation technique in the preparation of tissue reference materials for a range of shellfish toxins. Anal Bioanal Chem. 2007;387:2487–93.
Quilliam MA, Reeves K, MacKinnon S, Craft C, Whyte H, Walter J, Stobo L, Gallacher S (2006) Preparation of reference materials for azaspiracids. Proceedings of the 5th International Conference of Molluscan Shellfish Safety, 14–18 June 2004, Galway, Ireland. Molluscan Shellfish Safety, ISBN: 1 902895-33-9, pp. 111–115
McCarron P, Giddings SD, Quilliam MA. A mussel tissue certified reference material for multiple phycotoxins. Part 2: liquid chromatography-mass spectrometry, sample extraction and quantitation procedures. Anal Bioanal Chem. 2011;400:835–46.
McCarron P, Emteborg H, Giddings SD, Wright E, Quilliam MA. A mussel tissue certified reference material for multiple phycotoxins. Part 3: homogeneity and stability. Anal Bioanal Chem. 2011;400:847–58.
Doucet E, Ross NN, Quilliam MA. Enzymatic hydrolysis of esterified diarrhetic shellfish poisoning toxins and pectenotoxins. Anal Bioanal Chem. 2007;389:335–42.
ISO-Guide-30 (2015) Reference materials – selected terms and definitions. ISO/REMCO, Geneva, Switzerland
McCarron P, Kilcoyne J, Hess P. Effects of cooking and heat treatment on concentration and tissue distribution of okadaic acid and dinophysistoxin-2 in mussels (Mytilus edulis). Toxicon. 2008;51:1081–9.
Hess P, Nguyen L, Aasen J, Keogh M, Kilcoyne J, McCarron P, et al. Tissue distribution, effects of cooking and parameters affecting the extraction of azaspiracids from mussels, Mytilus edulis, prior to analysis by liquid chromatography coupled to mass spectrometry. Toxicon. 2005;46:62–71.
King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T. Mechanistic investigation of ionization suppression in electrospray ionization. J Am Soc Mass Spectrom. 2000;11:942–50.
Kilcoyne J, Fux E. Strategies for the elimination of matrix effects in the liquid chromatography tandem mass spectrometry analysis of the lipophilic toxins okadaic acid and azaspiracid-1 in molluscan shellfish. J Chromatogr A. 2010;1217:7123–30.
ISO-5725-2 I (1994) Accuracy (trueness and precision) of measurement methods and results. Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method. ISO, Geneva, Switzerland
Kilcoyne J, McCarron P, Twiner MJ, Nulty C, Crain S, Quilliam MA, et al. Epimers of azaspiracids: isolation, structural elucidation, relative LC-MS response, and in vitro toxicity of 37-epi-azaspiracid-1. Chem Res Toxicol. 2014;27:587–600.
Quilliam MA, Xie M, Hardstaff W. A rapid extraction and cleanup procedure for the liquid chromatographic determination of domoic acid in unsalted seafood. J AOAC Int. 1995;78:543–54.
Falk M, Walter JA, Wiseman PW. Ultraviolet spectrum of domoic acid. Can J Chem. 1989;67:1421–5.
ISO-17511 (2003) In vitro diagnostic medical devices-measurement of quantities in biological samples-metrological traceability of values assigned to calibrators and control materials. ISO, Geneva, Switzerland
Miller WG. Why commutability matters. Clin Chem. 2006;52:553–4.
Suzuki T, Beuzenberg V, Mackenzie L, Quilliam M. Liquid chromatography-mass spectrometry of spiroketal stereoisomers of pectenotoxins and the analysis of novel pectenotoxin isomers in the toxic dinoflagellate Dinophysis acuta from New Zealand. J Chromatogr A. 2003;992:141–50.
Acknowledgments
Technical assistance from Sabrina D. Giddings, Ruth A. Perez, Kelley L. Reeves, and William Hardstaff at the National Research Council is acknowledged. This project was supported by the Marine Institute (Galway, Ireland), the European Commission’s Joint Research Center (Geel, Belgium), the Norwegian Veterinary Institute (Oslo, Norway), and AgResearch (Hamilon, New Zealand). Dr. Philipp Hess (Ifremer, Nantes, France), formerly of the Marine Institute (Galway, Ireland), is acknowledged for his significant contributions to this project in its early stages.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 444 kb)
Rights and permissions
About this article

Cite this article
McCarron, P., Wright, E., Emteborg, H. et al. A mussel tissue certified reference material for multiple phycotoxins. Part 4: certification. Anal Bioanal Chem 409, 95–106 (2017). https://doi.org/10.1007/s00216-016-0004-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-0004-0