
Abstract
The earthworm represents a kind of creature in contact with the soil surface and usually exposed to a variety of organic pollutants from human activities. Therefore, it can be considered as an organism of choice for identifying pollution or better understanding the input of contaminants in food chains in particular through the contributions of sludge. Moreover, the use of organisms such as soil invertebrates is to be developed for ecotoxicological risk assessment of pollutants. In this context, a simple, rapid and effective multi-residue method was developed for the determination of 31 compounds including 11 steroids, 14 veterinary antibiotics and 6 human contaminants (paracetamol, sulfamethoxazole, fluvoxamine, carbamazepine, ibuprofen, bisphenol A) in earthworm. The sample preparation procedure was based on a salting-out extraction with acetonitrile (QuEChERS approach) that was optimised with regard to the acetonitrile/water ratio used in the extraction step, the choice of the clean-up and the quantity of the matrix. The optimised extraction method exhibited recoveries that comprised between 44 and 98 % for all the tested compounds. The limits of detection of all compounds were below 14 ng g−1 and the limits of quantification (LOQ) comprised between 1.6 and 40 ng g−1 (wet weight). The method was therefore applied to determine the levels of pharmaceuticals and hormones in six earthworm samples collected in various soils. Concentrations up to 195 ng g−1 for bisphenol A were determined, between a few nanograms per gram and 43.1 ng g−1 (estriol) for hormones and between a few nanograms per gram and 73.5 ng g−1 (florfenicol) for pharmaceuticals. Experiments were also conducted in laboratory conditions to evaluate the accumulation of the target substances by earthworm.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Soils are contaminated by emerging pollutants such as pharmaceutical and veterinary products or hormones primarily by diffusion in fields and by sludge from sewage treatment plants or manure and liquid manure produced in barns [1–4]. Indeed in the last 30 years, the application of manure to agricultural soils in several European countries has been a political orientation consisting of waste recovery while providing an effective way to fertilise the soil. Published data, however, indicate that, depending on the animal, between 10 and 90 % of veterinary drugs or hormones administered are excreted in urine or faeces as their non-metabolised form [1, 3].
Using chemical measurement in the soil matrices (soil water and particulate phase) is not sufficient to fully address the issue of identifying the causes of the disturbances or changes occurring in the ecosystem. The use of sentinel organisms belonging to the soil microfauna to assess soil contamination by emerging contaminants could be very relevant but is still uncommon. Some organisms such as earthworms are yet primary consumers of organic matter in the soil and are recognised to bioaccumulate minerals [5, 6] and organic contaminants, through soil consumption or contact. Recent studies showed the presence of contaminants from wastewater treatment plant sludge in earthworms living in amended soils [7, 8]. Concentrations well above the levels recorded in the corresponding soils confirm the earthworm’s potential for the bioaccumulation of organic micro-pollutants. Representing up to 60–80 % of the soil biomass [9], earthworms can be considered as organisms of choice for identifying points of input of contaminants in food chains in particular through the contributions of sludge.
On the other hand, the use of organisms such as soil invertebrates is to be developed for ecotoxicological risk assessment of pollutants (e.g. emerging pollutants) via evaluation of their transfer (bioavailability and bioaccumulation) and induced effects. Any disruption of retention and of soil habitat functions by contaminants can be studied with invertebrates such as earthworms by analysing the residues of these contaminants in their tissues. As these invertebrates are included in many food chains, the ecotoxicity data they provide would also be useful for addressing the risks associated with a trophic transfer. At times, it is not possible to link these levels of contaminants in the tissues with disruption or impacts at various key stages in their life cycle, or to identify the contaminant fraction actually available for transfer to earthworm and the potential induction of toxic effects, by the lack of analytical methods. Moreover, according to the VICH programme that aimed at harmonising technical requirements for veterinary product registration in the European Union and worldwide [10], the risk assessment of veterinary pharmaceuticals to soil organisms has to be considered. Thus, it is appropriate to develop methodologies for multi-residue analysis of veterinary compounds in earthworms.
Methods for quantifying some emerging micro-pollutants in earthworms are rare. The most difficult and time-consuming task for the determination of organic micro-pollutants in solid environmental matrices is the sample preparation, which often combines one or more extractions and purifications. Extractions are based on a simple solid-liquid extraction by an organic solvent [11, 12] or on using techniques of pressurised liquid extraction (PLE) [7] or assisted by ultrasounds [8, 13, 14] that reduce sample preparation time and the quantities of the solvent required. Purification is then carried out by solid-phase extraction (SPE) [7, 12] or by gel permeation chromatography [13, 14]. The selectivity and sensitivity required are then obtained by the development and optimisation of a separation by liquid chromatography (LC) coupled to detection by mass spectrometry (MS) or tandem mass spectrometry (MS/MS).
The limited data on quantification of pharmaceutical compounds in earthworms originates from the studies dealing with the accumulation of compounds from agricultural fields after biosolid or swine manure applications. Trimethoprim (antibiotic) was detected at levels of 61 and 127 ng/g dry weight in earthworms collected in fields amended by biosolid and manure, respectively [7]. Low concentrations of albuterol (bronchodilator) in biosolid-amended soil varied between 0.8 and 1.9 ng/g dry weight in a biosolid-amended soil, while the level of acetaminophen comprised between 74 and 100 ng/g dry weight in the same earthworms [15].
Recently, the so-called quick, easy, cheap, effective, rugged and safe (QuEChERS) extraction was developed by Anastassiades et al. to extract pesticide residues in plant materials mainly for food safety applications [16]. This method is based on a buffered salting-out extraction with acetonitrile followed by a dispersive solid-phase extraction for the clean-up. This method combines several significant advantages such as its simplicity, rapidity and low-solvent consumption. Therefore, this technology is of great interest and emerged these past 12 years. For these years, the two steps of this method have been optimised and adjusted for the extraction of various compounds (veterinary drugs, pesticides, pharmaceuticals, hormones) in different environmental matrices, including animal tissues [17, 18], honeybees [19], fishes [20, 21] or benthic invertebrates [22].
The objective of the present paper is to present the development of an innovative, rapid, simple, robust and sensitive method inspired by the QuEChERS method dedicated to the analysis of 11 steroids (androstenedione, testosterone, progesterone, norethindrone, gestodene, levonorgestrel, estriol, estrone, 17β- and 17α-estradiol, 17α-ethinylestradiol), 14 veterinary antibiotics (sulfanilamide, sulfadiazine, sulfathiazole, sulfametoxydiazine, trimethoprim, sulfadimerazine, sulfabenzamide, sulfadimethoxine, erythromycin, tylosin roxithromycin, penicillin G, dicyclanil, florfenicol) and 6 human contaminants (paracetamol, sulfamethoxazole, fluvoxamine, carbamazepine, ibuprofen, bisphenol A) in earthworms. This sample preparation is followed by a sensitive LC-MS/MS analysis. To our knowledge, the development of such approach for earthworm’s analyses has never been proposed.
Experimental section
Materials and reagents
High purity analytical standards were used, at least 98 % purity. Thus, sulfanilamide, sulfadiazine, sulfathiazole, sulfametoxydiazine, trimethoprim, sulfadimerazine, sulfabenzamide, sulfadimethoxine, sulfamethoxazole, dicyclanil, erythromycin, tylosin tartrate, penicillin G potassium salt, androstenedione, testosterone, norethindrone, levonorgestrel, progesterone, paracetamol, fluvoxamine, carbamazepine, bisphenol A, 17α- and 17β-estradiol, estrone, estriol, florfenicol and ibuprofen were obtained from Sigma-Aldrich (Saint Quentin Fallavier, France). The steroid gestodene was purchased from AK Scientific (CA, USA) and 17α-ethinylestradiol from Fluka. Only roxithromycin, furnished by Sigma-Aldrich, showed a purity of only 90 %.
Individual standard solutions were prepared in methanol (MeOH) at concentrations of 250 mg/L and stored at −20 °C for 6 months. Working standard mixtures were prepared by the appropriate mixture of the stock solutions and their dilution.
MeOH and hexane (HEX) of HPLC grade and acetonitrile (ACN) of LC-MS grade were obtained from Sigma-Aldrich. Ultra-pure water was obtained from a purification system Gradient A10 from Milli-Q (Saint-Quentin-en-Yvelines, France).
QuEChERS acetate buffers and dispersive solid-phase extraction (dSPE) phases were purchased from Agilent Technologies (Massy, France). The buffer composition was as follows: 6 g of anhydrous magnesium sulphate and 1.5 g of sodium acetate. Two phases of different compositions were tested for the dSPE clean-up. The first contained 950 mg of MgSO4 and 150 mg of primary and secondary amine-bonded silica (PSA), while the second contained 950 mg of MgSO4, 150 mg of PSA and 150 mg of C18-bonded silica (PSA/C18).
Sampling and sample preparation
Adult Canadian earthworms used for the development and validation were purchased from a fishing shop (Lyon, France). Prior to the assays, all earthworms were placed on wet filter paper for a minimum of 24 h, during which time they emptied their guts. After this depuration step, earthworms were cleaned using deionised water and stored at −20 °C until extraction and analysis.
For the bioaccumulation study, the same earthworms were used. Five kilograms of soil was collected from the region of Lyon and were spiked at 100 ng g−1 with a solution containing all the target compounds. The spiked soil was homogenised and placed in a sterile box. At the beginning of the study, which represented the zero time (t 0) and after three (t 1) and seven (t 2) days of exposure, four earthworms were removed, rinsed with pure water and kept in wet filter for 24–48 h to allow the gut to empty. Then, the earthworms were rinsed with pure water again and stored in a glass flask at −20 °C prior to the determination of pharmaceuticals.
For the application of the method to real samples, earthworms sampled from different locations around the Lyon conurbation were used. Site A corresponded to a corn field that received liquid cow manure as an organic fertiliser 2 months before the collection. Site B corresponded to a pasture that received liquid cow manure a week before the collection. Site C was a private garden not subjected to any outside stress. Sites D and E corresponded to a private kitchen garden receiving horse manure. This garden was tilled several months after application (site D) and 1 week after application (site E). Site F corresponded to a private home compost. For each site, about 10 earthworms were crushed together to achieve a good representation and then aliquots of 250 mg were used for analysis.
Sample extraction
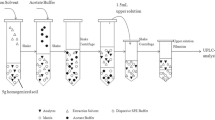
Before the QuEChERS procedure, earthworms were crushed and homogenised. Then, about 250 mg of homogenised earthworms were transferred into a 50-mL polypropylene tube. Volumes of 10 mL of ACN and 6 mL of water were added and then the mixture was shaken during 20 s with a vortex device (Vortex Fischer Scientific FB15013 TopMix). After that, a volume of 3 mL of hexane was added. The mixture was then swirled on a vortex mixer for an additional 40 s. The acetate buffer was then rapidly added and the tube was immediately manually shaken to avoid agglomeration for 20 s and swirled on the vortex mixer for 40 s. The tube was then shaken for 2 min at 1250 rpm in a sample homogeniser (SPEX Sample Prep, 2010 GenoGrinder, Delta Labo, Avignon, France). After centrifugation at 5000 rpm for 2 min (Sigma Laboratory Centrifuges 3K30H, Fisher Bioblock Scientific), 6 mL of the ACN layer was transferred into a 12-mL dSPE tube containing the dSPE phase (950 mg of MgSO4, 150 mg of PSA and 150 mg of C18). After that, the clean-up tube was manually shaken during 20 s and then swirled on a vortex mixer during 40 s. Finally, 4 mL of the purified extract was evaporated to dryness under a gentle stream of nitrogen at a temperature of 40 °C and finally reconstituted in 500 μL of H2O/MeOH (95/5; v/v) for LC-MS/MS analysis.
LC-MS/MS analysis
The liquid chromatographic separation was performed on an Agilent Series 1100 HPLC system from Agilent Technologies equipped with a degasser, a binary pump, an autosampler and a column oven. The separation in the positive ionisation mode was performed with a Zorbax Eclipse PLUS C18 (50 × 2.1 mm, 1.8 μm, Agilent Technologies) column preceded by a column pre-filter KrudKatcher from Phenomenex. The mobile phase was composed of 0.01 % formic acid in Milli-Q water (pH = 3.3) (A) and MeOH (B) with the following gradient: from 100 to 90 % (A) in 2 min, 90 % (A) for 2 min, from 90 to 80 % (A) in 3 min, from 80 to 71 % (A) in 1.80 min, 71 % (A) for 0.40 min and from 71 to 0 % (A) in 5.80 min. In the negative ionisation mode, the separation was performed with a Zorbax Eclipse XDB C18 (100 × 2.1 mm, 1.8 μm, Agilent Technologies) column preceded by the same column pre-filter. The mobile phase was composed of (A) Milli-Q water and (B) 50/50 ACN/MeOH with the following gradient: from 95 to 80 % (A) in 1.5 min, from 80 to 54 % (A) in 1.5 min, 54 % (A) for 2.40 min, from 54 to 20 % (A) in 11.40 min and from 20 to 0 % (A) in 0.60 min. Two different columns were chosen for both ionisation modes because three compounds had similar transitions (17α-estradiol, 17β-estradiol and estrone) and had to be separated. They were not separated by the Zorbax Eclipse PLUS C18 with the use of MeOH as organic solvent in the mobile phase. Only the addition of ACN led to a separation of these compounds but resulted in a decreased sensitivity for other compounds. Finally, to improve the separation, it was necessary to choose a Zorbax Eclipse XDB C18 in negative mode and to use a mixture of ACN/MeOH as organic solvent. For both separations, the column oven temperature was 50 °C, the flow rate was 0.3 mL/min and the injection volume was 25 μL.
The LC system was coupled to a triple-stage 3200 QTrap from AB Sciex (Les Ulis, France) with an electrospray ion (ESI) source (Turbo V, AB Sciex). The MS/MS settings and the parameters of the ESI source were optimised by manual infusion with a syringe pump and by the flow injection of standards, as described by Salvia et al. [23]. They are presented in Table 1. The source parameters were as follows: the ion source gas nebuliser was 45 and 40 psi in ESI+ and ESI−, respectively; the ion source turbo gas was 55 and 50 psi in ESI+ and ESI−, respectively; the ion spray voltage was 5500 and −4500 V in ESI+ and ESI−, respectively; and the source temperature was 600 and 500 °C in ESI+ and ESI−, respectively. The chromatographic conditions were previously optimised and discussed in [23]. The analytes were identified by both their chromatographic characteristics (comparison of the retention time with a standard; ±0.1 min) and their specific multiple reaction monitoring (MRM) fragmentation patterns (the presence of two characteristic transitions MRM1 and MRM2 as well as the compliance of the MRM1/MRM2 ratio with respect to standards, ±20 %). The stability of the LC-MS/MS instrument was assessed between runs by including a control and assessing the stability of the retention times (±0.1 min), the presence of MRM1 and MRM2 and the stability of the specific ratios of MRM1/MRM2 (deviation <20 %). Data processing was performed with Analyst software (version 1.5.1).
Validation
The whole method including the sample preparation based on QuEChERS and the LC-MS/MS analysis was validated using the International Conference of Harmonisation (ICH) guidelines [24]. The limits of detection (LOD) and quantification (LOQ) were defined as the compound concentration that produced a chromatographic peak signal 3 and 10 times the background noise, respectively.
The linearity was evaluated by analysing six earthworms spiked at six various concentrations of each of the compounds of interest, each of which was then extracted. The 6 points corresponded to 1 × LOQ, 2 × LOQ, 3 × LOQ, 4 × LOQ, 5 × LOQ and 10 × LOQ of the method. Linearity was considered validated if the determination coefficient on the concentration range (r 2) was superior than 0.99. To estimate the repeatability (or intra-day precision), the earthworm samples were spiked, extracted and analysed under the same conditions by the same manipulator and on the same day. The analysis of two levels of concentration was repeated three times. The intra-day precision is expressed as the relative standard deviation (RSD, %) of these measurements. Intermediate precision or inter-day precision was evaluated at the same levels of concentration as used for repeatability. In order to introduce variations in the process leading to the determination of intermediate precision, two analysts executed the process, different bottles of solvents were used and the procedures were realised over a period of 3 days. The intermediate precision was also expressed as the RSD of these measurements.
Results and discussion
The original QuEChERS method consists of a two-step extraction [16]. The first step is a liquid/liquid extraction using ACN as organic solvent that promotes the extraction of micro-pollutant residues. After shaking, anhydrous MgSO4 and NaCl are then added to promote the water partition from the organic phase and its dehydration. In order to make the QuEChERS method suitable for a wide group of matrices and contaminants, two buffers have rapidly been proposed instead of NaCl, based on acetate and citrate, respectively. An aliquot of the acetonitrile phase is recovered and then the second step consists in a dSPE clean-up. The sorbent phase interacts with previously co-extracted compounds, thus removing them from the acetonitrile phase.
The objective of the sample preparation of earthworms based on the QuEChERS approach was to allow an optimal extraction of all the targeted compounds while minimising the presence of interfering substances. The extraction procedure is based on a weight-to-volume ratio. To respect these ratios, it was necessary to adapt the proportion of the organic solvent to the matrix weight. Consequently, optimisation of the QuEChERS extraction was achieved by assessing three experiments: (i) the mass of the earthworm sample, (ii) the acetonitrile/water volume ratio (V ACN/V water) and (iii) the purification step. To optimise each step, the samples were spiked at 100 ng g−1.
Earthworm’s matrix mass
The mass of the earthworm was the first parameter to be studied. Three different masses were tested, i.e. 250, 500 and 1000 mg of the matrix that were extracted by QuEChERS. Noteworthy, no purification was used during this optimisation. The optimisation of the mass was based on the recovery (R) between samples spiked before (S −) extraction and the extract obtained by the extraction of the matrix and spiked after extraction (S +) by the following equation: R % = (S − / S+) × 100.
Recoveries for the different masses are given in Fig. 1. Overall, samples of 250 mg exhibited higher recoveries, especially for dicyclanil, penicillin G, carbamazepine and estriol. For instance, an average recovery of 65 % was computed for substances spiked in samples of 250 mg, while 45 and 37 % was obtained for average recoveries at 500 and 1000 mg, respectively. The extractions from 250 mg showed a higher uncertainty than from 500 to 1000 mg, i.e. up to 25 % for roxithromycin and sulfanilamide, while the maximum uncertainties for 500 and 1000 mg were 18 (sulfanilamide) and 13 % (carbamazepine), respectively. Overall, a smaller sample quantity coupled to a higher solvent to sample ratio probably favours the extraction efficiency of the target compounds and minimises the extraction of competitive interfering compounds. The smallest amount, i.e. 250 mg, exhibited the highest recovery and was used as the optimised parameter for all the subsequent experiments.

Recoveries measured as a function of the initial mass of earthworm (V ACN = V water = 10 mL, no clean-up)
V ACN/V water ratio for extraction
The liquid-liquid extraction is based on the affinity of the compound for each phase. This affinity is primarily dependent on the compound, then on the nature and/or the polarity of the solvents and finally on the volume ratio of solvents used during extraction. Therefore, the V ACN/V water ratio was optimised. According to the original QuEChERS approach [16], a 10-g sample (mainly composed of water) is introduced in a tube with 10 mL acetonitrile, i.e. a ratio of 1/1 (w/v) for promoting the extraction of residues. To enhance the extraction of target compounds, the relative amount of ACN was increased compared to the original method. Taking into account the water content of the earthworms (80 %), V ACN/V water ratios of 3.1, 1.6 and 1.0 were subsequently tested. The results are presented in Fig. 2. Note that regardless of the ratio, yields comprised between 75 and 100 % for antibiotics, except penicillin G (50 %). Recoveries and almost relative standard deviation were much more variable with other target substances. The best compromise to achieve good recoveries while ensuring good repeatability of extraction was the ratio 1.6. It was therefore used for all forthcoming extractions of targeted substances.

Recoveries and matrix effects measured as a function of the V ACN/V water ratio (initial mass = 250 mg, no clean-up)
Clean-up
The matrix effects represent a common phenomenon in the analytical methods based on mass spectrometry. They correspond to enhancement or inhibition of analytical signals and, thus, either positively or negatively influence the ionisation of the analytes. These matrix effects are due to matrix compounds that can be eluted at the same retention times as the compounds of interest. The matrix effects are therefore dependent on the nature of the matrix-interfering compounds and the recovery of the sample preparation step. Therefore, the QuEChERS method implies a clean-up that should eliminate interfering compounds without removing the analytes of interest. As the earthworms represent a complex matrix, it was important to thoroughly study this phenomenon.
To evaluate this matrix effect (ME), two signals were compared. The first corresponds to the signal of the earthworm extracted and then spiked with all the target substances (A extract). The second corresponds to the signal of standards at the same concentration in solvent (A solvent). The percentage of ME was then calculated according to the following equation:
Therefore, several clean-ups were evaluated by comparison of both the recovery of the target compounds and the diminution of ME. This clean-up was performed by dSPE that allows the retention of co-extractants of the matrix but not the targeted substances, on the dSPE sorbent. To selectively trap the matrix-interfering substances, different dSPE sorbents exist, such as PSA or the mixture PSA/C18. The PSA sorbent is a weak anion exchanger usually used to eliminate polar organic acids, sugars and lipids, whereas the PSA/C18 is efficient to remove less polar compounds. In both cases, the addition of MgSO4 eliminates the water content in the extract and consequently enhances the partition of the interfering substances on the sorbent. Figure 3 resumes the recoveries with both clean-up steps and Fig. 4 the matrix effects.

Recoveries measured without clean-up and with PSA or PSA/C18 clean-up (initial mass = 250 mg, V ACN = 10 mL, V water = 6 mL)

Matrix effects measured without clean-up and with PSA or PSA/C18 clean-up
In terms of extraction, the use of a PSA phase decreased overall recoveries. Decreases by a factor of 2 to 3 were observed for the acidic substances (e.g. ibuprofen). The loss was lower with the use of the PSA/C18; the same recovery was obtained for most substances. The presence of both phases probably creates a hydrophilic-lipophilic balance beneficial to the passage of the target compounds in the acetonitrile phase. In addition, the use of a PSA/C18 improved the repeatability of the sample preparation compared to the method without purification. In terms of cleaning efficiency, the signal suppressions were generally reduced with the use of a dispersive phase. An increase of the signal with the use of the PSA phase was noteworthy. Overall matrix effects were weaker in the presence of PSA/C18. Using this phase coupled to PSA was previously shown to be a more efficient clean-up in fatty food matrices [25]. The maximum ion suppression was computed for fluvoxamine (−86 %), roxythromycin (−77 %) and gestoden (−64 %). Overall, no correlation between ionisation mode and matrix effects was observed. Indeed, substances ionised in the positive mode presented similar average matrix effects than those ionised in the negative mode (average suppression of 29 and 28 %, respectively). To resume, the dSPE phase composed of 950 mg of MgSO4, 150 mg of PSA and 150 mg of C18 was retained. This clean-up step decreases the signal suppression due to matrix effects and, therefore, improves the sensitivity of the method. In addition, the diminution of untargeted co-extracted compounds in the system lengthens the lifetime of the chromatographic column and reduces the fouling of the mass spectrometer.
Method validation and method performance
Validation is recognised to be a crucial step, as it proves the consistency of an analytical method which allows its application to real samples. The criteria used for validation were the LOD, LOQ, linearity, recovery, repeatability and intra- and inter-day precisions (Table 2). It should be noted that some hormones were naturally present in earthworms at various concentrations. Therefore, it was impossible to get a matrix free of the target hormones. In consequence, to obtain a homogenous sample for conducting the validation plan, 20 earthworms were crushed together. To determine the amount of the hormones naturally present in this pool, the non-spiked pool was extracted on each day of the validation. All the spiked and non-spiked samples were obtained from this same sample pool.
Table 2 shows the results for the validated linearity range of the method. For all the compounds except ibuprofen, the determination coefficients (r 2) were superior than 0.99 in this range. The contraceptives gestodene and 17α-ethinylestradiol exhibited a narrower range of linearity (40–8000 ng g−1).
Recoveries comprised between 45.2 % (sulfamethoxazole) and 105 % (erythromycin) at the LOQ level and between 33.7 % (fluvoxamine) and 115 % (17α-estradiol) at 20 × LOQ (Table 2). The recovery values depended on both the compound and the concentration, but averages were respectively 78 and 74 % at concentrations corresponding to LOQ and 20 × LOQ, respectively. For the LOQ spiking level, recoveries can be classified into three groups. Group A represents molecules with recovery <60 % (19 % of the total of analytes), group B those with recovery between 60 and 80 % (35 %) and group C those molecules presenting recovery >80 % (46 %). Thus, sulfathiazole, sulfadiazine, sulfamethoxazole, carbamazepine and androstenedione were included in group A; they displayed the lowest median recoveries. The second group consisted of dicyclanil, sulfabenzamide, roxythromycin, paracetamol, testosterone, norethindrone, gestodene, bisphenol A and estriol. Meanwhile, the other compounds were recovered in earthworms at median recoveries lying between 85 and 105 %. It was not possible to compare the performances of the method developed with previous published works that used QuEChERS extraction because, to our knowledge, it is the first time that this method is used for the extraction of steroids, veterinary antibiotics or other human contaminants from earthworm. The works published by Kinney et al. [7] represent probably the only multi-residue and multi-family approach dealing with the analysis of emerging micro-pollutants in earthworms. In this paper, PLE followed by SPE as clean-up was used for the analysis of organic anthropogenic waste indicators including three pharmaceutical compounds and bisphenol A. The authors reported recoveries between 33 and 117 %, which is globally comparable to the performance of our method. The recovery of the antiepileptic carbamazepine achieved using our method was lower than that obtained by Kinney et al. [7]. On the other hand, the latter did not allow achieving high recovery for bisphenol A, unlike the QuEChERS method with which we reached recovery of 75 or 88 %, depending of the level.
LOQ values (Table 2) comprised between 1.6 and 40 ng g−1. The values vary within the same chemical family. For example, in the case of steroids, the LOQ of estrone was 1.6 ng g−1, whereas that of 17α-ethinylestradiol was 40 ng g−1.
With regard to intra-day precision, good repeatability was obtained, with RSD that comprised between 2.9 and 8.7 % when evaluated at the level corresponding to LOQ (Table 2). RSD was inferior to 15 % at the level corresponding to 20 × LOQ, except testosterone (56.3 %). Inter-day precisions were also very good because they were inferior to 10 % regardless of the compounds.
It is also interesting to note that three molecules do not satisfy the ICH validation, i.e. sulfanilamide, penicillin G and ibuprofen. For sulfanilamide and penicillin, the method did not exhibit reproducible extractions because yields varied when the level corresponded to the LOQ. Regarding ibuprofen, r 2 coefficient was less than 0.90, which makes it impossible to confirm the linearity within the range tested and did not allow quantifying the substance.
Application to real samples
The QuEChERS-LC-MS/MS method developed in this study was applied to earthworms sampled from six different locations around the Lyon conurbation. The results are resumed in Table 3 and the chromatograms obtained from the analysis of the earthworms of site E are presented in Fig. 5 as an illustration. The results indicated that 27 of the target molecules were detected or quantified at least once in the samples. Among them, five (namely fluvoxamine, testosterone, progesterone, levonorgestrel and bisphenol A) were detected in 100 % of the collected samples, and among them, three were systematically quantified (testosterone, levonorgestrel and bisphenol A). The presence of testosterone and progesterone in earthworms from the unstressed sample can probably be at least partially explained by their natural presence in earthworms. This hypothesis was confirmed by the relatively close levels measured between different samples. On the other hand, the synthetic progestogen levonorgestrel and the antidepressant fluvoxamine were quantified in all the samples with higher levels in sites E and F. The presence of bisphenol A results from its extensive use in industry and household applications, which leads to a greater release into the environment. This phenomenon is highlighted by comparing the levels measured between unstressed (C) or low-stressed samples (A, B, D) with kitchen garden (E) and compost (F). Levels are 4–8 times lower in the less stressed sites. The levels of veterinary substances range from less than LOQ up to several dozen nanograms per gram.

Total ion current (TIC) chromatograms of earthworms collected in site E submitted to the whole protocol. ESI+: (1) sulfanilamide, (2) paracetamol, (3) dicyclanil, (4) sulfadiazine, (5) sulfathiazole, (6) sulfamethoxydiazine, (7) sulfadimerazine, (8) sulfamethoxazole, (9) sulfabenzamide, (10) sulfadimethoxine, (11) carbamazepine, (12) roxithromycin, (13) erythromycin, (14) androstenedione, (15) testosterone, (16) levonorgestrel and (17) progesterone. ESI−: (1′) florfenicol, (2′) estriol, (3′), bisphenol A, (4′) estrone and (5′) ibuprofen
The results indicate that earthworms are exposed to a large number of organic micro-pollutants through the application of manure or compost. The method developed in this study was also applied to follow the uptake of the target compounds in laboratory conditions. This operation did not aim to establish the rate of absorption contaminants by earthworms or their kinetics but whether they can be considered as biological tracers of the environmental contamination by the target compounds. Thus, a soil sample was spiked with all of the compounds, and the substance concentrations in the earthworms were measured at the beginning and then after 3 and 7 days.
The results are summarised in Table 4. For most of the substances, concentrations after 7 days of exposure were higher than those for 0 and 3 days of exposure. This predominance highlights the importance of the absorption pathway for pharmaceuticals in earthworms.
Note that several substances were not detected, even after 7 days. Yet some of them were quantified in samples collected in environmental conditions in soil (Table 3). This was the case of dicyclanil, sulfadimethoxine, fluvoxamine, and 17α- and 17β-estradiol. This could probably come from the time of the experiment, too short for the absorption of the molecule by the earthworm. Another hypothesis could be a metabolisation of the substances by the worm. Indeed, it is known that the regulation of endogenous steroid levels in molluscs is controlled by both stereogenesis and steroid metabolism [26]. On the other hand, the metabolisation of pharmaceutical substances has already been discussed in a risk assessment study [27].
As regards sulfadiazine and estriol, concentrations were initially increasing before decreasing. Further works would be needed to better understand this phenomenon, but first, hypotheses could be a biotransformation and/or a link with bioavailability of substances within the soil compartment.
Conclusion
An innovative, selective and effective method was implemented for the determination of hormones and pharmaceuticals in earthworms. The multi-residue method consisted of a sample preparation protocol based on the buffered salting-out extraction named QuEChERS followed by a selective and sensitive LC-MS/MS analysis. This extraction is simple and rapid, does not require expensive equipment and involves only a low-solvent consumption, which is increasingly put forward in the era of sustainable development. Therefore, this technology brings many benefits not only in analytical aspect but also in toxicological, environmental and economic levels.
This methodology was successfully applied to the analysis of earthworms collected in unstressed or amended soils, as well as in compost. Experiments conducted in laboratory conditions also highlighted the potential of accumulation of the target compounds. These first experiments showed that earthworms represent good sentinel species of the terrestrial ecosystem and could therefore be useful as a tracer of human activities on soil and to better understand the input of these pollutants through food chains.
References
Kumar K, Gupta SC, Chander Y, Singh AK (2005) Antibiotic use in agriculture and its impact on the terrestrial environment. In: Advances in Agronomy, volume 87, Elsevier, pp 1–54
Johnson A, Jurgens M (2003) Endocrine active industrial chemicals: release and occurrence in the environment. Pure Appl Chem 75:1895–1904
Shore LS, Shemesh M (2003) Naturally produced steroid hormones and their release into the environment. Pure Appl Chem 75:1859–1871
Diercxsens P, Tarradellas J (1987) Soil contamination by some organic micropollutants related to sewage sludge spreading. Int J Environ Anal Chem 28:143–159
Suthar S, Singh S (2009) Bioconcentrations of metals (Fe, Cu, Zn, Pb) in earthworms (Eisenia fetida), inoculated in municipal sewage sludge: do earthworms pose a possible risk of terrestrial food chain contamination? Environ Toxicol 24:25–32
Suthar S (2008) Metal remediation from partially composted distillery sludge using composting earthworm Eisenia fetida. J Environ Monit 10:1099–1106
Kinney CA, Furlong ET, Kolpin D, Burkhardt MR, Zaugg SD, Werner SL, Bossio JP, Benotti MJ (2008) Bioaccumulation of pharmaceuticals and other anthropogenic waste indicators in earthworms from agricultural soil amended with biosolid or swine manure. Environ Sci Technol 42:1863–1870
Macherius A, Lapen DR, Reemtsam T, Römbke J, Topp E, Coors A (2014) Triclocarban, triclosan and its transformation product methyl triclosan in native earthworm species four years after a commercial-scale biosolids application. Sci Total Environ 472:235–238
Bouche A (1992) In: Greig-Smith PW, Becker H, Edwards PJ, Heimbach F (eds) Ecotoxicology of earthworms. Intercept, Andover, pp 20–35
VICH (International Cooperation on Harmonization of Technical Requirements for registration of Veterinary Medicinal Products), GL (2004) Environmental impact assessments (EIAs) for veterinary medicinal products—phase II. CVMP/VICH/790/03. European Medicine Evaluation Agency, London
Carter LJ, Garman CD, Ryan J, Dowle A, Bergström E, Thomas-Oates J, Boxall ABA (2014) Fate and uptake of pharmaceuticals in soil-earthworm systems. Environ Sci Technol 48:5955–5963
Zhao S, Zhu L, Liu L, Liu Z, Zhang Y (2013) Bioaccumulation of perfluoroalkyl carboxylates (PFCAs) and perfluoroalkane sulfonates (PFSAs) by earthworm (Eisenia fetida) in soil. Environ Pollut 179:45–52
Wang H, Chen J, Guo B-Y, Li J (2014) Enantioselective bioaccumulation and metabolization of diniconazole in earthworms (Eisenia fetida) in an artificial soil. Ecotoxicol Environ Saf 99:98–104
Chen J, Wang H-L, Guo B-Y, Li J-Z (2014) LC-MS/MS method for simultaneous determination of myclobutanil, hexaconazole, diniconazole, epoxiconazole and tetraconazole enantiomers in soil and earthworms. Int J Environ Anal Chem 94:791–800
Kinney CA, Campbell BR, Thompson R, Furlong ET, Kolpin DW, Burkhardt MR, Zaugg SD, Werner SL, Hay AG (2012) Earthworm bioassays and seedling emergence for monitoring toxicity, aging and bioaccumulation of anthropogenic waste indicator compounds in biosolids-amended soil. Sci Total Environ 433:507–515
Anastassiades M, Lehotay SJ, Štajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86:412–431
Stubbings G, Bigwood T (2009) The development and validation of a multiclass liquid chromatography tandem mass spectrometry (LC-MS/MS) procedure for the determination of veterinary drug residues in animal tissue using a QuEChERS (QUick, Easy, CHeap, Effective, Rugged and Safe) approach. Anal Chim Acta 637:68–78
Abdallah H, Arnaudguilhem C, Jaber F, Lobinski R (2014) Multiresidue analysis of 22 sulfonamides and their metabolites in animal tissues using quick, easy, cheap, effective, rugged, and safe extraction and high resolution mass spectrometry (hybrid linear ion trap-Orbitrap). J Chromatogr A 1355:61–72
Wiest L, Bulete A, Giroud B, Fratta C, Amic S, Lambert O, Pouliquen H, Arnaudguilhem C (2011) Multi-residue analysis of 80 environmental contaminants in honeys, honeybees and pollens by one extraction procedure followed by liquid and gas chromatography coupled with mass spectrometric detection. J Chromatogr A 1218:5743–5756
Norli HR, Christiansen A, Deribe E (2011) Application of QuEChERS method for extraction of selected persistent organic pollutants in fish tissue and analysis by gas chromatography mass spectrometry. J Chromatogr A 1218:7234–7241
Orlando EA, Simionato AVC (2013) Extraction of tetracyclinic antibiotic residues from fish filet: comparison and optimization of different procedures using liquid chromatography with fluorescence detection. J Chromatogr A 1307:111–118
Berlioz-Barbier A, Buleté A, Faburé J, Garric J, Cren-Olivé C, Vulliet E (2014) Multi-residue analysis of emerging pollutants in benthic invertebrates by modified micro-quick-easy-cheap-efficient-rugged-safe extraction and nanoliquid chromatography-nanospray-tandem mass spectrometry analysis. J Chromatogr A 1367:16–32
Salvia MV, Vulliet E, Wiest L, Baudot R, Cren-Olivé C (2012) Development of a multi-residue method using acetonitrile-based extraction followed by liquid chromatography-tandem mass spectrometry for the analysis of steroids and veterinary and human drugs at traces level in soil. J Chromatogr A 1245:122–133
(2005) ICH Harmonised Tripartite Guideline, validation of analytical procedures: text and methodology Q2(R1). International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use
Bruzzoniti MC, Checchini L, De Carlo RM, Orlandini S, Rivoira L, Del Bubba M (2014) QuEChERS sample preparation for the determination of pesticides and other organic residues in environmental matrices: a critical review. Anal Bioanal Chem 406:4089–4116
Fernandes D, Loi B, Porte C (2011) Biosynthesis and metabolism of steroids in molluscs. J Steroid Biochem 127:189–195
Boleas S, Alonso C, Pro J, Mar Babin MM, Fernandez C, Carbonell G, Tarazona JV (2005) Effects of sulfachlorpyridazine in MS 3-arable land: multispecies soil system for assessing the environmental fate and effects of veterinary medicines. Environ Toxicol Chem 24:811–819
Acknowledgments
The authors thank the ADEME (Agence de l’Environnement et de la Maîtrise de l’Energie) for the ARCOS programme funding (no. 10 94 C 0076).
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
The authors declare that the experiments were conducted in accordance with animal care.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bergé, A., Vulliet, E. Development of a method for the analysis of hormones and pharmaceuticals in earthworms by quick, easy, cheap, effective, rugged and safe (QuEChERS) extraction followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Anal Bioanal Chem 407, 7995–8008 (2015). https://doi.org/10.1007/s00216-015-8972-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8972-z