
Abstract
Highly portable, cost-effective, and rapid-response devices are required for the subtyping of the most frequent food-borne bacteria; thereby the sample rejection strategies and hygienization techniques along the food chain can be tailor-designed. Here, a novel biosensor is presented for the generic detection of Salmonella and Campylobacter and the discrimination between their most prevalent serovars (Salmonella Enteritidis, Salmonella Typhimurium) and species (Campylobacter jejuni, Campylobacter coli), respectively. The method is based on DNA microarray developed on a standard digital versatile disc (DVD) as support for a hybridization assay and a DVD driver as scanner. This approach was found to be highly sensitive (detection limit down to 0.2 pg of genomic DNA), reproducible (relative standard deviation 4–19 %), and high working capacity (20 samples per disc). The inclusivity and exclusivity assays indicated that designed oligonucleotides (primers and probes) were able to discriminate targeted pathogens from other Salmonella serovars, Campylobacter species, or common food-borne pathogens potentially present in the indigenous microflora. One hundred isolates from meat samples, collected in a poultry factory, were analyzed by the DVD microarraying and fluorescent real-time PCR. An excellent correlation was observed for both generic and specific detection (relative sensitivity 93–99 % and relative specificity 93–100 %). Therefore, the developed assay has been shown to be a reliable tool to be used in routine food safety analysis, especially in settings with limited infrastructure due to the excellent efficiency-cost ratio of compact disc technology.

DNA microarray performed by DVD technology for pathogen genotyping
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Food-borne zoonotic diseases are an important human health problem in most countries, in spite of the improvement in hygiene tools during food processing and handling. Hundreds of thousands cases are reported each year in the European Union and USA [1, 2]. Food safety actions have been implemented including risk management (e.g., legislative measures) and risk assessment (e.g., data collection). Salmonella and Campylobacter are among the most important food-borne pathogens in the world, and consequently, specific regulations have been established. Consequently, controls are being increasingly applied in order to tailor the disinfection methods to targeted microorganisms and to minimize the risk for consumers of ingesting contaminated food (directly or by cross-contamination). Conventional methods detect these pathogens by enrichment and isolation on selective media [3, 4]. Specific morphology of colonies can be recognized, but confirmation and identification of the presumptive positive cultures require subsequent serological and biochemical tests. At least three antibody-antigen reactions are performed to identify a particular Salmonella serovar (Kauffmann–White scheme). In case of Campylobacter, further identification to the species level requires assays such as antibiotic sensitivity and biochemical tests (sodium hippurate hydrolysis reaction). These laborious methods take several days to complete (3 to 5 days for Salmonella [3] and 4 to 8 days for Campylobacter [4]) and often lead to an erroneous identification of Campylobacter species. Besides, the possible simultaneous presence of several pathogens, species, and serotypes increase the number of assays required for each food product. There are a special interest in the simultaneous detection of both pathogens Salmonella and Campylobacter, discriminating the presence of their most prevalent serovars (Salmonella Enteritidis, Salmonella Typhimurium) or species (Campylobacter jejuni, Campylobacter coli).
The advances on proteomics and genomics allowed the research community to have molecular fingerprinting of food-borne pathogens, identifying specific proteins and genes for distinct lineages and sub-lineages within a bacterial population [5]. Using available information, different bioanalytical methods have been developed [6, 7].
DNA-based methods, particularly PCR methods, have experienced a huge growth in the last years due to their excellent performances. In fact, the real-time PCR technique (qPCR) is considered as the gold standard method for testing microbial contamination. In the last years, a new generation of methods has been proposed, incorporating properties such as miniaturization, portability, and cost-effectiveness [8, 9]. The reported methods for bacteria screening include electrochemical biosensors [10], lab-on-a-chip devices [11, 12], and chemiluminescence-based reactors [13], among others. However, the main drawback of some methods is the restricted number of targets that can be analyzed per reaction, i.e., the limited choices of fluorescent detectors in qPCR. Nucleic acid microarrays, also called gene chips, provide a robust system for the simultaneous detection of several microbial pathogens [14–17]. Conventional approaches are based on hybridization of fluorescent-labeled amplified products to an array of oligonucleotide probes immobilized on a glass support covalently [18].
However, the intrinsic properties of microarray scanners (size, price, and maintenance) restrict the use of this technology to centralized laboratories, frequently far from the sampling locations or food industry needs. In this way, our research group has recently shown the advantages of audio-video technology-based biosensors for testing microbial contamination in low-level labs [19, 20]. These systems integrate compact discs as polymeric supports for carrying out assays in microarray format and standard compact disc drives as detectors. The methodology involves similar steps than those based on glass slide microarrays, but the detection of target/probe biorecognition is performed by an advanced, very simple, inexpensive, and portable optical sensing system.
In this study, efforts have been focused on setting up of an assay based on digital versatile disc (DVD) technology for identification of prominent serovars of Salmonella sp. and species of Campylobacter. Important challenges have been addressed to overcome some of the disadvantages of described techniques. First, the oligonucleotide design was critical to amplified and detect all targeted bacteria, present in very low amounts among a background of indigenous microflora and other abundant serovars (e.g., S. Infantis). Second, as food products are of perishable nature, the screening methodology has been developed to be completed in the shortest time and highest multiplexing capability. Third, an upper level of demonstration has been reached by analyzing poultry products in an inter-laboratory validation study.
Experimental
Bacterial strains and growth conditions
Reference bacterial strains (targeted and non-targeted serovars and organisms), supplied by CECC (Spain), NCTC (UK), and ATCC (USA), are listed in Table 1. Salmonella strains were aerobically grown at 37 °C in ASAP medium (BioMerieux, Marcy l’Etoile, France). Campylobacter was grown in mCCDA medium (Oxoid, Basingstoke, UK) at 42 °C in microaerophilic conditions. After overnight incubation from fresh colonies, bacterial cells were collected and subjected to genomic DNA isolation.
Meat samples
Chicken samples bought in local supermarkets were inoculated using serial dilutions (100 to 105 CFU/mL) of pure culture (S. Enteritidis, S. Typhimurium, C. jejuni, and C. coli).
Sample collection was carried out in slaughterhouse facility focusing on broiler batches and broiler carcasses to determine the degree of microbial contamination at the beginning of the process line. This critical location was selected because contamination, growth, and survival of targeted microorganisms could occur if the intervention or preventive strategy were not working effectively or operations must be carried out or corrected by staff [21]. The campaign included 170 samples, being analyzed by microbiological identification and qPCR. For the inter-laboratory validation assay, a hundred samples were determined by DVD-based microarray method following a randomized controlled trial (blind samples).
The enrichment of pathogens used 25 g of each sample placed in a stomacher bag containing 225 ml of Buffered Peptone Water (Oxoid) for Salmonella and Preston Broth (Oxoid) for Campylobacter. After 20-h and 24-h incubation, respectively, a mixture of both samples was subjected to qPCR and disc-based microarray analysis. The microbiological identification of targeted pathogens was achieved by individual culturing in ASAP agar and xylose-lysine-deoxycholate agars for Salmonella and mCCDA medium for Campylobacter, after enrichment during 20 and 48 h, respectively. Bacteria classification was based on the analytical profile index (API, BioMerieux). Quantitative real-time PCR analysis (qPCR) was performed under an external certified laboratory (ISO17025) conditions. Four amplification reactions were prepared per sample: Salmonella sp. and Campylobacter sp. in single format, and S. Enteritidis/S. Typhimurium and C. coli/C. jejuni in duplex format, including specific probes conjugated minor grove binders. The detection limits of these standardized qPCR methods were 1 CFU/25 g of food.
DNA isolation and quantification
For microarray analysis, genomic DNA was extracted from 0.2 ml of bacterial culture (peptone water and Preston broth) using the automatic method Maxwell16 (Promega, Madison, WI) following the manufacturer’s instructions. The quantity and quality of the purified DNA was determined using a BioPhotometer (Eppendorf, Germany) by measuring A260 and the ratio A260/A280, respectively.
Design of multiplex PCR primers and microarray probes
Gene targets used were selected for generic detection of Salmonella and Campylobacter, as well as their serotypes or species, respectively. Histone-like protein Hlp-II gene (hns gene) and rRNA-16S ribosomal RNA gene (16S gene) were selected for generic detection of Salmonella and Campylobacter, respectively. Threonine operon leader peptide/predicted phage protein gene (sdf gen) and putative cytoplasmatic protein gene (STM4497 gene) were selected for specific identification of serovars S. Enteritidis and S. Typhimurium, respectively. Hippurate hydrolase gene (hipO gene) and enterochelin uptake periplasmic binding protein gene (ceuE gene) were chosen for specific identification of C. jejuni and C. coli species, respectively.
Specific oligonucleotides (two PCR primers and one probe) were designed for microarray analysis, totally independent to primers of qPCR method. The nucleotide sequences were chosen from those registered in the GenBank database of National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov). The Primer 3Plus software (http://www.bioinformatics.nl) was used to design all primers. Probes were designed in the unique sequence regions of these targeted genes, checking the absence of stable secondary structures or self-complementarity. A tail of 10 thymines was included in order to reduce the surface interactions. For quality control of the immobilization/developing process, a 40-mer oligonucleotide (5′-digoxigenin-T15-GTCATGGGCCTCGTGTCGGAAAACC-Biotin-3′) and a 35-mer oligonucleotide (5′-biotin-T10-TAGAGACTTAAAGAGGGAGCCCGGG-3′) were chosen as positive and negative probes, respectively. No sequence homology was found in any microbial genomes (BLAST search). All oligonucleotides used were synthesized by Eurofins (Ebersberg, Germany).
Spotting of probes on DVD chips
Bulk DVDs were purchased from MPO Iberica (Madrid, Spain). Probes (100 nM) prepared in printing solution (190 nM streptavidin, 50 mM carbonate buffer, pH 9.6, and 1 % glycerol (v/v)) were transferred to the disc (50 nL) with a non-contact arrayer (AD 1500 BioDot, Inc., Irvine, CA). The working temperature and relative humidity were controlled at 25 °C and 90 %, respectively. The layout was 7 × 9 spots per array and 20 arrays per DVD, being the distances between flanking spots of 1.5 mm. Hence, each array has nine replicate spots corresponding to each gene (hns, sdf, STM4497, 16S, hipO, and ceuE genes), two positive controls, and four negative controls (immobilization and hybridization). The spot diameter was 500 ± 10 μm. Complementary experiments were performed to determine the immobilization density, based on the measurement of fluorescence produced after the printing/incubation of a double-labeled oligonucleotide (5′-Cy5-T10-TTTGATTACAGCCGGTGTACGACCCT-Biotin-3′).
Hexaplex PCR
The reaction mixture (10 μL) was prepared adding 1.5 U Taq polymerase, 2 mM MgCl2, 200 μM dNTPs, and 20 μM digoxigenin-labeled dUTPs to the reaction buffer. The primer concentrations used for asymmetric amplification were 0.1 μM/0.4 μM (forward/reverse). The amount of DNA template per reaction was 1 ng for pure cultures and 5–10 ng for meat samples. A negative (human genomic DNA) and a positive control (mixture of genomic DNA from pure cultures of S. Typhimurium and C. coli) were included in each experimental batch. The thermocycling was carried out in a TC400 thermocycler (Bibby Scientific, Staffordshire, UK) as follows: 95 °C for 5 min for initial activation; 40 cycles of denaturing at 94 °C for 30 s, annealing at 56 °C for 30 s, and extension at 72 °C for 30 s; and 72 °C for 5 min for a final extension. The amplification factor was calculated from the number of copies synthesized and the amount of genomic DNA added to reaction solution, considering that the length of bacteria genome is 4760 kb for Salmonella (4.9 fg DNA/cell) and 1640 kb for Campylobacter (1.7 fg DNA/cell). The size of PCR products was determined by electrophoretic separation in 3 % (w/v) agarose gels and visualization by fluorescent staining.
Hybridization and scanning of DVD chips
All reagents were directly dispensed on disc using a silicon gasket of 20 wells, simplifying the working protocol. A fraction of the amplification product (3 μL) was mixed with 57 μL of hybridization buffer (saline sodium citrate, NaCl 650 mM, sodium citrate 65 mM, 25 % formamide pH 7). The solution was denatured at 95 °C for 5 min and chilled on ice, then applied onto the surface area of an array which was pre-framed grid in silicon sheet (Electron Microscopy Sciences, Hatfield, England). Discs were introduced inside a container (common plastic box for DVDs) at water-saturated atmosphere, and the hybridizations carried out at 37 °C for 45 min into a heating oven. Discs were gently washed with washing buffer (NaCl 15 mM, sodium citrate 1.5 mM, pH 7) and water. The immobilized product reacted with a mixture of sheep anti-digoxigenin antibody at 1:4,000 dilution (Dig-Ab) and rabbit horseradish peroxidase-labeled anti-sheep antibody (HRP-Ab) at 1:500 dilution in phosphate buffer saline (PBS-T, phosphate 100 mM and NaCl 1.5 M, plus 0.05 % Tween 20 at pH 7.5). Discs were incubated during 25 min at room temperature and darkness. For the developing step, 1 mL of 3,3′,5,5′-tetramethylbenzidine solution (TMB, Sigma-Aldrich, St. Louis, MO) was added and incubated 8 min at room temperature and darkness.
The optical densities of microarrays were directly read by the DVD drive (rotation speed 4× ≡ 13.46 m/s, 26 dB gain, 1700 Mega-samples/s), being the reading time lower than 10 min. Gray-scale images (tagged image file format, color depth 16 bit, scale 0–65,535) were analyzed. Complete image processing (feature gridding, addressing, segmentation, quality assurance) was automatically performed using in-home software in less than 5 min by disc. The mean intensity of each spot was calculated from 450 pixels with the highest intensity (equivalent spot diameter 200 μm).
Results
Optimization of the multiplex PCR
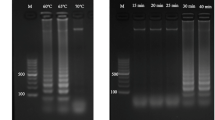
A hexaplex PCR was developed to simultaneously amplify all targeted genes using specifically designed primers (Table 2). The optimization set included genomic DNA extracts from several Salmonella and Campylobacter strains. The most important variables to achieve a high amplification factor were found to be MgCl2 concentration, number of cycles, and annealing temperature. Then, a three-factor experiment design was applied, indicating the optimum values as follows: 2 mM, 40 cycles, and 56 °C, respectively. The optimization of primer concentrations, following a six-factor experimental design, indicated that the primers of hns and 16S genes (generic detection of both pathogens) should be added in a lower concentration to obtain similar amplification factors for the six targeted genes. Probably, these variations were sequence-associated due to the formation of secondary structures. Also, asymmetric amplification was studied modifying the primer concentrations from 1:1 to 1:100 (forward/reverse). Variation of the ratio between both primers induced a linear amplification producing differences in the replicated amounts of each strand and rendering double- and single-strand DNA. This strategy helped latter hybridization to immobilized probes on DVD platform. The best results were achieved using a 1:4 ratio, being the higher concentration for the primer (reverse complementary strand) with respect to the probe. Under the selected conditions (Table 2), six targeted genes were specifically amplified (factors between 2.7 × 109 and 2.5 × 1010). In all cases, the products obtained had the predicted size (Fig. 1).

Agarose gel electrophoresis image of hexaplex PCR products. Pathogen concentration 16 × 103 CFU/μL. Fluorescent dye: real safe. Lane 1, 50 bp mini DNA ladder (Fisher Scientific International Inc.). Lane 2, S. Typhimurium CECT 433. Lane 3, S. Enteritidis CECT 4300. Lane 4, Salmonella Nottingham NCTC 7832. Lane 5, C. jejuni ATCC 33560; lane 6, C. coli ATCC 33559. Lane 7, Cronobacter sakazakii ATCC BBA-894. Lengths of PCR products, 139 bp (hns gene), 189 bp (sdf gene), 62 bp (STM4497 gene), 108 bp (16S gene), 85 bp (hipO gene), and 81 bp (ceuE gene)
Optimization of microarray detection
Layout
Amplification products from multiplex PCR were hybridized on the bottom layer of DVD discs. The microarray layout was selected considering the intrinsic properties of the substrate surface and the optical resolution of the DVD reader. For a conventional disc (12 cm diameter), the available sensing area starts at a radius of 2 cm and finish at 5.9 cm (track pitch area), being approximately 97 cm2. On the other hand, the optical resolution of the DVD driver used, as microarray scanner, is about 8.2 μm/pixel (laser diode emission λ = 650 nm, numerical aperture NA = 0.6). Given these high performances of DVD technology, dozens of probes/arrays can be potentially immobilized allowing the detection of a high number of different samples or identify more bacterial types by assay. As proof-of-concept, a low-density format, based on silicon gasket, was chosen for detecting 20 independent samples/replicates in a single assay. The microarray dimensions were 10.5 × 13.5 mm (7 × 9 spots, inter-flanking distance 1.5 mm), occupying 2.3 % of DVD area per microarray (including inter-sample barriers). The use of low-volume printing systems (few nL), lower inter-flanking distance between spots, or other compartmentation systems (e.g., drops, hybridization chambers) can improve the number of microarrays per disc [20]. The multiplexing capabilities, expressed by number of samples/assay, are better than conventional planar chips. Therefore, massive screening in food safety applications can be addressed using the proposed method.
Immobilization strategy of probes
The proposed hybridization assay involved the immobilization of biotin-labeled probes via streptavidin adsorption on raw surface of standard DVDs (bottom layer, grooved polycarbonate substrate with track pitch). The selected option is quite simple compared to covalent attachment of probes described for glass or plastic substrates [22]. From the practical point of view, the maximum immobilization density, approximately 0.1 fmol/mm2, was reached incubating 190 nM streptavidin and 100 nM probes at pH = 9.6 during 12 h at 4 °C. The system took advantage of the demonstrated compatibility of streptavidin-biotin recognition (affinity constant K d ≈ 10−15 M) and the simultaneous protein physisorption on disc. These results obtained using double-labeled probes were also confirmed by the later hybridization of PCR products at fixed concentration.
Hybridization
Multiplex PCR products were hybridized on printed probe microarray, by dispensing a dilution in stringent buffer. No post-amplification steps were required except a fast denaturation by heating. The chosen format avoided time-consuming and expensive strategies such as labeling by Klenow polymerase or purification in silica columns [23, 24]. Reactions were carried out at 37 °C for 45 min into a simple heating oven. An immunoassay combined with an enzymatic enhancement was selected as developing reaction, as shown in Fig. 2a. As digoxigenin-labeled nucleotides were added during the amplification process, the products were specifically recognized by the combination of antibodies. Optimization experiments indicated that the best dilutions were 1:4000 for primary antibody and 1:500 for secondary antibody (Fig. 2b). Due to the hydrophobic properties of polycarbonate, no blocking of DVD surface was needed to avoid unspecific binding of reaction mixtures.

a Illustration of developing reaction: digoxigenin-labeled PCR products are recognized by sheep anti-digoxigenin antibody and then by the anti-sheep antibody conjugated with rabbit horseradish peroxidase. The addition of a substrate generates a solid deposit on the spot, where the specific target DNA-probe complex is formed. b Optical intensity of spot depending on the concentration of developing antibodies: PCR product from a pure culture of S. Typhimurium CECT 443
Detection
The DVD drive reading principle was based on the variation of the reflection properties of the DVD surface due to the presence of the biorecognition product. During the disc scanning, the reflected laser reached the photodiode of the pickup, generating the background signal (maximum reflection). But, when the laser hit a microarray spot, an attenuation of the reflected beam is produced, and consequently, the intensity of laser beam decreased. Figure 3 images are generated after surface scanning of each array, registering signal-to-noise ratios up to 30. Spots were unambiguously distinguished from background, determining six genes in a single array. The spot intensity was related to the amount of PCR product incubated on the microarray.

a Scheme of reading principle by a DVD drive: the reflections of the laser beam following the spiral track are converted into electrical pulses and collected by data acquisition card. As the laser starts reading the disc from the inside ring and ends up on the outside, the data from the complete disc is registered and the software generates an image by microarray. b Microarray layout: inter-spot distance 1.5 mm, spot diameter 0.55 mm. C−: negative control, non-complementary probe; C+: positive control, digoxigenin-labeled probe. c Microarray images obtained for pure cultures (pathogen concentration 16 × 103 CFU/μL)
Analytical performances
The sensitivity was examined by hybridizing the multiplex PCR products from a series of 10-fold diluted genomic DNA. Yet, the mixtures contained from 0.01 pg to 10 ng of extracted DNA from each targeted strain. The lowest amounts of genomic DNA detected as positive responses (signal-to-noise ratio higher than 3) were 1.6 ± 0.6 × 10−4 ng for Salmonella and 1.7 ± 0.6 × 10−4 ng for Campylobacter. Sensitivity was also determined applying the method to 10-fold diluted pure cultures, being the detection limit 14–57 CFU/mL for Salmonella and 11–60 CFU/mL for Campylobacter, prior to DNA extraction and PCR amplification. For pure cultures, a clear relationship between optical density captured by DVD drive and pathogen concentration was observed (Fig. 4). Sensitivity variations observed among the different probes should be due to the discrepancy of product concentrations (amplification efficiency) or differences in hybridization yield. A 100-fold increase of detection sensitivity was reached compared to single PCR and subsequent gel electrophoresis. This improvement is mainly due to the selected enzymatic development of microarrays. The assay reproducibility was calculated from culture extracts with 1 ng/μL of genomic DNA from each pathogen. Intra-assay reproducibility, expressed as the relative standard deviation of three replicates, performed in the same assay, was 4–19 %. Inter-assay reproducibility, expressed as the relative standard deviation of three replicates, performed in different assays, was 8–24 %.

Standard curves for reference pathogens in pure cultures: a S. Enteritidis; b S. Typhimurium; c C. jejuni; d C. coli
The assay selectivity was evaluated in terms of the inclusivity (detection of the target organism) and exclusivity (nondetection of nontarget microorganisms). Table 3 summarizes the results obtained analyzing a selected set of pure cultures (pathogen concentration 2⋅105 CFU/mL). Negative and positive controls provided the expected responses, supporting the quality assurance of the hexaplex PCR and hybridization assay. The microarray-based method succeeded in detecting all serovars of Salmonella tested (total of 21 isolates belonging to 19 serovars) and identifying the targeted serotypes. In case of detection of Campylobacter, neither false-positive nor false-negative were reported for pure culture isolates analyzed (10 isolates). Genomic extracts from reference strains of other pathogens were assayed to determine the exclusivity (13 isolates). No response was observed in any case.
Regarding to the analysis time, starting from a minimal bacterial cells, genomic DNA extraction (1 h, <2 €/sample) and PCR amplification (nearly 2 h, <1.5 €/sample) are general protocols of microbial DNA methods. The specific steps of DVD-based method are performed in 2 h: hybridization (70 min), developing reaction (40 min), DVD reading (5 min), and image processing (2 min), being the hands-on-time about 30 min. The estimated cost of materials and reagents is less than 1 € per disc (20 samples).
Application to food safety control
DVD microarray detection was applied to identify the presence of Salmonella and Campylobacter in meat samples bought in local supermarkets and poultry products collected in a slaughterhouse facility.
The analysis of inoculated chicken samples indicated that the method provides discrepancies with respect to the spiked amount. Probably, the matrix effect affects the polymerase activity, changing the amount of PCR products at the end of the amplification reaction. These phenomena have been described for techniques based on end-point PCR [25]. Then, the proposed approach was applied as semiquantitative method in meat samples (concentration degrees).
In the slaughterhouse facility, the sample collection covered the processing chain from raw material to fresh poultry meat preparations. An inter-laboratory validation study was designed in order to demonstrate the capabilities of the proposed method. Hence, a set of 100 blind samples were determined by DVD microarray approach. Thirteen samples provided negative responses for all targets. The presence of Salmonella or Campylobacter was detected in 21 and 67 samples, respectively. Respect to the specific identification, S. Enteritidis (10 cases) was more detected than S. Typhimurium (9 cases) or other Salmonella serovars (9 cases). C. jejuni was identified in 52 cases, C. coli in 13 cases, and both in 2 cases being the only Campylobacter species detected. The information was used for the elaboration of effective tailor-made actions against these human health hazards, i.e., specific hygienization.
Microarray results were compared to the obtained by microbiological/biochemical methods and quantitative real-time PCR (single/duplex format) to the same insolates. Figure 5 shows an example of the results obtained in poultry samples with microbiological contamination. The relative sensitivity and specificity of DVD-based method were calculated from the number of positive and negative results, according to reference methods. The relative sensitivity for generic detection was 95.5 % for Salmonella (one case of false-negative) and 98.5 % for Campylobacter (one case of false-negative). The relative specificity was 100 % for both pathogens (no false-positives). Also, a successful specific identification of Salmonella serovars (sensitivity 94.4 % and selectivity 98.9 %) and Campylobacter species was reached (sensitivity 92.9 % and selectivity 96.2 %). Although the number of meat samples analyzed is limited (n = 100), the results obtained by microarray detection agreed well with the reference figures, demonstrating the reliability of the DVD-based technique for screening purposes. The multiplex approach was especially interesting because raw meat is a common source and vehicle for transmitting all these virulent strains to humans [6–8]. Hence, the microarray method provided an important reduction of time analysis and reagent consumption as well as a better high-throughput capacity (e.g., number of samples simultaneously analyzed) than most of current methods [3, 4, 8, 9, 26].

Results for 25 poultry samples with positive presence of pathogens. Microarray results are shown in a color scheme based on optical densities registered by DVD drive: green—negative, light red—positive with a low signal (concentration <102), red—positive with an intermediate signal (concentration 102–104), dark red—positive with a high signal (concentration >104). Results of reference methods (qPCR and microbiological/biochemical analysis) are expressed as positive presence (+) or negative presence (−)
The studied method also provides similar or improved performances than other microarray-based assays for detecting food-borne pathogens. For instance, recent oligonucleotide microarray methods have been developed with detection limits of 1–200 CFU per 25 g sample [10, 17–20, 23, 24, 27–29]. The proposed tool has important advantages over the current array methods, such as an economical (500–2000-folds cheaper detector), easy-to-use diagnostic tool (DVD technology), and fully automated data analysis for a high-throughput use. Also, the pathogens panel (including serovars) and/or the samples number analyzed on a disc in parallel may be gradually expanded through addition of newly designed probes or microarrays into the system. In fact, thousands of 500-μm-diameter dots can be printed on the DVD surface.
Regarding to the food safety control of meat products, this approach is able to screen products along the entire production/distribution chain with best performances than methods based on integrated devices or real-time PCR (few targets) and DNA microarrays (expensive instruments). The proposed device had improved properties like versatility, portability, and low cost of acquisition and maintenance. After using conventional enrichment culture-based techniques, the isolates were identified by a portable, rapid, reliable, and cost-effective method. Therefore, the developed system fulfills the requirement for an alternative microbiological testing during production and processing [6, 7].
Conclusions
An extensive monitoring of food safety, especially microbial contamination, involves the use of reliable, speed-to-answer, and cost-effective methods. However, a vast majority of food companies do not have access to robust, effective, and quick-response microbiological techniques. Exploiting a consolidated consumer electronics as DVD technology, a powerful biosensing tool is achieved and validated. First, the device covered the demand of simple, robust, low-cost instrument/platform without compromising detection capabilities. Second, the developed methodology satisfies the expected analytical performances. In this sense, the present study demonstrates that it is possible to identify targeted serovars and species in a single assay with high sensitivity and specificity (closed to 100 %). Their better performances than RT-PCR-based methods, particularly cost and working capability, will support an enhanced control of food-borne pathogens and the effective management of hygiene measurements. Hence, this novel approach can be easily integrated in a hazard analysis and critical control points (HACCP) system, adapting the production lines and accelerating the distribution of perishable foods.
References
European Food Safety Authority (2014) EFSA J 12:3547
Gould LH, Walsh KA, Vieira AR, Herman K, Williams IT, Hall AJ, Cole D (2013) Morb Mortal Wkly Rep (MMWR) 62:1–34
ISO 6579:2002, Microbiology of food and animal feeding stuffs. Horizontal method for the detection of Salmonella spp
ISO 10272–1:2006, Microbiology of food and animal feeding stuffs. Horizontal method for detection and enumeration of Campylobacter spp. Part 1: detection method
Kim S, Frye JG, Hu J, Fedorka-Cray PJ, Gautom R, Boyle DS (2006) J Clin Microbiol 44:3608–3615
Jasson V, Jacxsens L, Luning P, Rajkovic A, Uyttendaele M (2010) Food Microbiol 27:710–730
Park SH, Aydin M, Khatiwara A, Dolan MC, Gilmore DF, Bouldin JL, Ahn S, Ricke SC (2014) Food Microbiol 38:250–262
Lazcka O, Del Campo FJ, Munoz FX (2007) Biosens Bioelectron 22:1205–1217
Velusamy V, Arshak K, Korostynska O, Oliwa K, Adley C (2010) Biotechnol Adv 28:232–254
Wang Y, Ye Z, Ying Y (2012) Sensors (Basel) 12:3449–3471
Mairhofer J, Roppert K, Ertl P (2009) Sensors (Basel) 9:4804–4823
Hsieh K, Patterson AS, Ferguson BS, Plaxco KW, Soh HT (2012) Angew Chem Int Ed 51:4896–4900
Donhauser SC, Niessner R, Seidel M (2011) Anal Chem 83:3153–3160
Vora GJ, Meador CE, Stenger DA, Andreadis JD (2004) Appl Environ Microbiol 70:3047–3054
Wang XW, Zhang L, Jin LQ, Jin M, Shen ZQ, An S, Chao FH, Li JW (2007) Appl Environ Microbiol 76:225–233
Lopez-Campos G, Martínez-Suarez JV, Aguado-Urda M, Lopez-Alonso V (2012) Microarray detection and characterization of bacterial foodborne pathogens. Springer, New York
Suo B, He Y, Paoli G, Gehring A, Tu SI, Shi X (2010) Mol Cell Probes 24:77–86
Gronlund H, Riber L, Vigre H, Lofstrom C, Folling L, Huehn S, Malorny B, Radstrom P, Rudi K, Hoorfar J (2011) Int J Food Microbiol 145:S79–S85
Arnandis-Chover T, Morais S, Tortajada-Genaro LA, Puchades R, Maquieira A, Berganza J, Olabarria G (2012) Talanta 101:405–412
Santiago-Felipe S, Tortajada-Genaro LA, Morais S, Puchades R, Maquieira A (2015) Food Chem 174:509–515
Jacxsens L, Kussaga J, Luning PA, Van der Spiegel M, Devlieghere F, Uyttendaele M (2009) Int J Food Microbiol 134:113–125
Sethi D, Gandhi RP, Kuma P, Gupta KC (2009) Biotechnol J 4:1513–1529
Kim HJ, Park SH, Lee TH, Nahm BH, Kim YR, Kim HY (2008) Biosens Bioelectron 24:238–246
Guo D, Liu B, Liu F, Cao B, Chen M, Hao X, Feng L, Wang L (2013) Appl Environ Microbiol 79:3392–3399
Smith CJ, Osborn AM (2009) FEMS Microbiol Ecol 67:6–20
Cheung PY, Kam KM (2012) Food Res Int 45:802–808
Feng J, Wang X, Cao G, Hu S, Kuang X, Tang S, You S, Liu L (2013) Eur Food Res Technol 236:1073–1083
Suo B, He Y, Irwin P, Gehring A (2013) Food Anal Methods 6:1477–1484
Shin HH, Hwang BH, Seo JH, Cha HJ (2014) Appl Environ Microbiol 80:366–373
Acknowledgments
This work has been supported by the Spanish Ministry of Economy and Competitiveness projects:INNPACTO (Ref. IPT-2011-1011-31000) and FEDER-CTQ2013-45875-R). Contribution of grant PROMETEO II/2014/040 (Generalitat Valenciana) was highly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tortajada-Genaro, L.A., Rodrigo, A., Hevia, E. et al. Microarray on digital versatile disc for identification and genotyping of Salmonella and Campylobacter in meat products. Anal Bioanal Chem 407, 7285–7294 (2015). https://doi.org/10.1007/s00216-015-8890-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8890-0