
Abstract
This paper describes a rapid and sensitive method for the determination of eugenol in fish samples, based on solid-phase extraction (SPE) and gas chromatography–tandem mass spectrometry (GC–MS–MS). Samples were extracted with acetonitrile, and then cleanup was performed using C18 solid-phase extraction (SPE). The determination of eugenol was achieved using an electron-ionization source (EI) in multiple-reaction-monitoring (MRM) mode. Under optimized conditions, the average recoveries of eugenol were in the range 94.85–103.61 % and the relative standard deviation (RSD) was lower than 12.0 %. The limit of detection (LOD) was 2.5 μg kg−1 and the limit of quantification (LOQ) was 5.0 μg kg−1. This method was applied to an exposure study of eugenol residue in carp muscle tissues. The results revealed that eugenol was nearly totally eliminated within 96 h.

Flow diagram for sample pretreatment
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Anesthetic is a drug that can inhibit the function of the central nervous system of animals [1]. Partial or whole-body anesthetic can be used on fish; after the use of anesthetic, the affected area will temporarily lose the feeling of pain and stress reaction will be reduced. Anesthetic has been widely used on fish in contexts including surgery, breeding, transport, and slaughter [2, 3].
Eugenol is a major constituent of the volatile oil of clove, and has been widely used in fish anesthesia [4–6]. The presence of eugenol in fish-derived food could pose a potential risk to human health because it may cause skin allergy and damage to the liver [7]. Because of these potential hazards, the eugenol residue in aquatic products has been the subject of increasing attention. The Joint Food and Agriculture Organization/World Health Organization (FAO/WHO) Expert Committee on Food Additives (JECFA) has established the acceptable daily human intake (ADI) of eugenol at 2.5 mg kg−1 body weight [8]. In Japan, eugenol is the licensed anesthetic for use on aquatic products and the maximum residue limit (MRL) is set at 50.0 μg kg−1 [9].
We are presently interested in studying the drug residue of eugenol in fish and evaluating the safety of eugenol residue. To achieve this, it is necessary to develop a precise and accurate analytical method for the analysis of eugenol level in fish samples. Many methods have been developed to determine eugenol in different matrices, including thin-layer-chromatography (TLC) densitometry combined with liquid–solid extraction in clove [10], high-performance liquid chromatography (HPLC) combined with liquid–solid extraction in defatted soybean flour [11], gas chromatography–tandem mass spectrometry (GC–MS–MS) combined with soxhlet extraction in fish [12], liquid chromatography–quadrupole ion-trap mass spectrometry (LC–QIT) combined with liquid–liquid extraction in rat plasma [13], and high-performance liquid chromatography (HPLC) combined with solid-phase extraction (SPE) in water [14].
Kildea et al. have developed a soxhlet extraction method combined with GC–MS–MS to determine eugenol in the edible tissue of silver perch, and have obtained interesting results [12]. However, soxhlet extraction usually takes several hours. Meinertz et al. have done some excellent work relating to eugenol, and have developed a SPE method to determine the eugenol exposure conditions that maximize the concentration of eugenol in rainbow-trout skin-on fillet tissue [14, 15]. However, the conditions of SPE still need to be studied in detail. In this study, we wanted to develop a simple and reliable method using SPE combined with GC–MS–MS to determine eugenol in fish.
In recent years, GC–MS–MS coupled with SPE has progressed to become the method of choice for fish bioanalysis [16, 17]. Eugenol has a phenol group and is a fat-soluble compound with pKa = 10.19 at 25 °C. Therefore, the use of SPE with a non-polar stationary phase can provide substantial benefits. The objective of our study was to develop a reliable, faster, and accurate SPE-GC–MS–MS method to determine eugenol in the muscle of fish. In the exposure experiment, living fish were exposed to eugenol solution and the residue level was determined by the developed method.
Experimental
Reagents and apparatus
The analytical standard eugenol (purity >97 %) was obtained from Chromadex (Muirlands Blvd, USA). Liquid-chromatography-grade acetonitrile and methanol were obtained from Merck (Darmstadt, Germany). Ultra-pure water was obtained from a Milli-Q system (Millipore Corporation, MA, USA). C18 SPE cartridges (300 mg, 3 mL) were obtained from Bestown (Beijing, China). Standard stock solutions of eugenol (100 mg kg−1) were prepared in pure acetonitrile. Standard working solutions at concentrations of 1–1000 μg L−1 were prepared from the stock solution by serial dilution. All solutions were stored in a refrigerator at −20 °C until use.
A homogenizer (IKA, Germany), a centrifuge (Xiangyi Corp., Hunan, China), and a vortex mixer (IKA, Germany) were used during sample preparation. A SPE instrument for 20 cartridges was obtained from Agilent Technologies (Santa Clara, CA, USA). A nitrogen-blowing evaporator was purchased from Organomation Associates, Inc. (MA, USA). Extracts were filtered through a polytetrafluoroethylene syringe filter with a pore size of 0.22 μm, purchased from Anpel Scientific Instrument Co. Ltd. (Shanghai, China).
Sample preparation
The living carp were obtained from a local market (Beijing, China). Muscle tissues were taken for analysis. They were first homogenized in a homogenizer and then stored at −20 °C until analysis. The weighed homogenized sample (2.0 g) was placed into a 50 mL centrifuge tube. A standard solution (100 μL) was added to the blank sample for recovery studies to validate the method. The sample was vortexed for 1 min, and placed at room temperature for 10 min. Then 10 mL acetonitrile was added, followed by vortexing for 1 min. The tubes were capped and centrifuged for 5 min at 4000 rpm at 4 °C. The supernatant was transferred into a 50 mL centrifuge tube. The extraction procedure was repeated and the two supernatants were merged. n-Hexane (10 mL) was added to defat. After mixing well by vortex, the tube was centrifuged at 4000 rpm for 5 min. The layer of n-hexane was discarded, and the defatting pretreatment process was repeated. The solution of acetonitrile was concentrated under nitrogen stream at 45 °C, until it became dry. Residue was redissolved with 5.0 mL methanol–water (1:9, v/v) and was then ready for purification.
Solid-phase extraction
C18 solid-phase-extraction cartridges were preconditioned with 3.0 mL methanol followed by 3.0 mL water. After equilibrating, 5.0 mL sample extract was loaded onto the clean cartridges. The cartridges were then washed sequentially with 3.0 mL acetate buffer (pH 4.0) and eluted with 3.0 mL methanol. The eluate was collected, and concentrated under a nitrogen stream. The concentrated residue was redissolved in 1.0 mL acetonitrile, filtered through a 0.22 μm polytetrafluoroethylene syringe filter, and then 2.0 μL of the solution was injected into the GC–MS–MS system.
Fish-exposure experiment
The exposure study was performed by short-term exposure to eugenol solution following the method described in our previous work [3]. Exposure baths were prepared in aquariums (160 × 60 × 80 cm, length × width × depth) which were filled with 10.0 mg L−1 eugenol solution. The living carp (approximately 200 g wet weight) were purchased from a market (Wuhan, China). GC–MS–MS was used to verify that no eugenol residue was present on the fish. The carp (n = 50) were transported to the laboratory and transferred into an aquarium exposure bath at 23 ± 1 °C. The fish were then transported to fresh water after 2 h. The levels of eugenol were detected at different intervals.
GC–MS–MS analysis
GC separation was performed using a 7693 series autosampler interfaced to a 7890A Series GC with a split/splitless injector (Agilent Technologies, Santa Clara, CA, USA). A 7000 series triple-quadrupole mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with an electron-ionization detector (EI) source in the multiple-reaction-monitoring (MRM) mode was used to analyze eugenol. The analytical column was a 30 m HP-5 capillary column (30 m × 0.25 mm × 0.25 μm). Samples were injected in the pulse splitless mode. The GC injector and transfer line were at a temperature of 280 °C. The ion-source temperature was also set to 280 °C. Helium was used as carrier gas and the flow was 1.0 mL min−1. The oven temperature program began at 70 °C (held for 1 min), was raised to 120 °C at 40 °C min−1, and then increased to the final temperature of 170 °C at 10 °C min−1 (held for 1 min).
Results and discussion
Optimization of GC–MS–MS
For the GC separation of eugenol, the use of a weakly polar stationary phase was found to be efficient. An HP-5 column (30 m × 0.25 mm × 0.25 μm) was used for the separation of eugenol. The eugenol produced a sharp peak, and the compound was quantified with peak-area integration. The GC–MS–MS chromatogram of eugenol is shown in Fig. 1. The mass spectrum of eugenol obtained by electron ionization at 10 eV is shown in Fig. 2. A parent ion for eugenol was observed as a molecular ion (M+) at m/z 164, and product ions also appeared at m/z 149, 137, 131, 121, 104, and 77. The quantification and confirmation ion transitions are listed in Table 1.

(a) GC–MS–MS chromatogram of standard eugenol solution at a concentration of 100 μg L−1; and (b) an extraction from a carp muscle tissue spiked at 50 μg kg−1

Electron-ionization mass spectrum obtained from a standard solution of eugenol at 500 μg L−1 (The marked ion is the molecular ion of eugenol)
Optimization of the extraction procedure
The analysis of complex matrices, including fish, needs rigorous sample preparation to obtain a stable and sensitive analysis. The log octanol–water partition coefficient (log Kow) of eugenol is 2.3 at pH 7, 20 °C. The extraction method is based on the principle of liquid extraction. Organic solvents enable the extraction of eugenol from fish samples, and simultaneously reduce background interference. The weighed homogenized sample (2.0 g) was placed into a 50 mL centrifuge tube. Standard solution (100 μL, 1000.0 μg L−1) was added to the blank sample for the optimization of extraction-solvent type. The sample was vortexed for 1 min, and then 10 mL extraction solvent was added followed by vortexing for 1 min. The tube was capped and then centrifuged at 4000 rpm for 5 min. The supernatant was transferred into a 15 mL centrifuge tube and concentrated under nitrogen stream at 45 °C. The residue was redissolved with 1 mL acetonitrile. In this study, two kinds of extraction solvent (acetonitrile and methanol) were investigated. The results revealed that higher extraction recoveries were obtained when acetonitrile was used as extraction solvent.
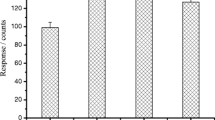
Then the volume of acetonitrile was evaluated, as illustrated in Fig. 3. When the volume of acetonitrile was 20 mL, the recoveries of eugenol were within a higher range. Therefore 20 mL acetonitrile was chosen as extraction solvent in the following experiments.

Optimization of the extraction solvent of spiked fish-muscle samples (50 μg kg−1)
Optimization of SPE process
To detect eugenol in fish samples at trace levels, cleanup using SPE is necessary. Eugenol is a fat-soluble compound, with pKa = 10.19 at 25 °C. Therefore, the use of a non-polar stationary phase was found to be effective. A C18 SPE cartridge (300 mg) was selected for the cleanup of eugenol. The type and volume of eluent solvent, which could affect SPE performance, were optimized. Three replicates were performed for these studies.
Effect of the type of eluent solvent
The type of eluent solvent is an important factor affecting the SPE efficiency of the analyte. In this study, four kinds of eluent solvent (acetonitrile, ethyl acetate, methanol, and n-hexane) were investigated. Acetonitrile extractions of spiked fish-muscle samples (50 μg kg−1) were concentrated under nitrogen stream and redissolved with 5 mL methanol–water (1:9, v/v) to obtain a sample solution. The sample solution was loaded onto the C18 SPE cartridge. After washing with 3 mL acetate buffer, the cartridge was eluted with 3 mL of the potential eluent solvent. The eluate was collected and concentrated under a nitrogen stream. The concentrated residue was redissolved in 1 mL acetonitrile for detection. As shown in Fig. 4, n-hexane obtained relatively low recovery, and methanol obtained the highest extraction recoveries. Therefore, methanol was chosen as eluent solvent in the following experiments.

Effect of eluent solvent on the recovery of eugenol attached to SPE cartridges
Effect of the volume of eluent
The volume of eluent is another important factor that affects the SPE efficiency of the analyte. Therefore, the effect of methanol volume on the extraction efficiency of eugenol was investigated. As shown in Fig. 5, the extraction recoveries reached the maximum value with 3 mL methanol as eluent solvent. With further increases of the volume of methanol, the recoveries remained almost constant. On the basis of these results, 3 mL methanol was enough to elute the retained eugenol and was therefore used in the following experiments.

Effect of the volume of methanol on the recovery of eugenol from samples spiked at 50 μg kg−1
Stability of eugenol in fish samples
The stability of eugenol in fish samples was investigated. The fish samples were spiked at 50 μg kg−1 and stored under different conditions. Two kinds of stability were investigated. The short-term stability was determined after 4 h storage at room temperature. The long-term stability was evaluated after one day or two days of storage at −20 °C. The results regarding stability are shown in Table 2. The short-term stability and long-term stability revealed, respectively, that eugenol was stable in fish samples at room temperature for 4 h and at −20 °C for at least 48 h.
Matrix effects
The matrix background of fish samples is complex. This causes some difficulties regarding accurate quantification of drug residues in fish samples. Investigation of matrix effects is needed for validation of quantitative determination for supporting exposure studies. To evaluate the matrix effect, chromatographic peak areas of eugenol obtained from the spiked samples after extraction were compared with those obtained from the standard solution at the same concentrations in neat solvent. The assessment was performed at low, medium, and high concentration levels. The matrix effects determined were 109.84 ± 12.39 %, 103.08 ± 7.26 %, and 105.11 ± 3.31 % at 25, 50, and 100 μg kg−1, respectively, for fish samples (Table 3). Minor enhancement of eugenol signal was observed as a result of endogenous matrix interferences including fatty acids and proteins, and had no effect on the quantification of eugenol.
Method validation
The SPE cleanup and the optimization of instrumental settings were evaluated in the method validation, which assessed sensitivity, linearity, and mean recovery.
The sensitivity was evaluated by determining the limit of detection (LOD) and the limit of quantification (LOQ).
The linearity was determined by injecting six standard solutions into GC–MS–MS, ranging from 1.0 to 500.0 μg L−1. An external calibration curve was achieved in the range 5.0 to 500.0 μg L−1 and the correlation coefficient (R 2) of the linear regression was >0.9931. Recoveries were calculated at each spiked level using the working standard calibration curve. The LOD and LOQ were calculated from spiked fish muscles on the basis of a signal-to-noise ratio (S/N) of 3.0 and 10.0, respectively. The LOD and LOQ were 2.5 μg kg−1 and 5.0 μg kg−1, respectively (Table 3). Three concentration levels were selected to test the recoveries over a specific range. The lower spiked concentration was selected to include the MRL concentration of 50.0 μg kg−1 wet weight set by the Japanese Positive List System for Agricultural Chemical Residues in Foods [9]. The blank samples were proved to be free of interferences by GC–MS–MS. Six replicates of the spiked fish muscles at the three different levels were prepared. The precision under these conditions of repeatability, expressed as the relative standard deviation (RSD), was determined (Table 3). All recoveries were satisfactory, with mean values ranging from 94.85 to 103.61 %, and RSD below 12.0 %. The recoveries of this study met the requirement (80.0–110.0 % for above 10.0 μg kg−1) of the Commission Decision 2002/657/EC at all three spiking levels [18]. The results suggested that the developed sample-preparation method coupled with GC–MS–MS was reliable and sensitive for quantifying eugenol in fish.
Fish-exposure experiment
The developed sample-preparation method was applied to the exposure study of eugenol. The fish were first transferred into 10.0 mg L−1 eugenol solution for 2 h, and then the levels of eugenol in the muscle tissues were detected (Table 4). Eugenol was eliminated quickly in muscle tissues, and nearly totally eliminated within 96 h.
Conclusions
This work reports a SPE cleanup method for the determination of eugenol residue in fish. Extracts were analyzed and validated using GC–MS–MS with an EI source. The recovery percentages were satisfactory, in the range 94.85 to 103.61 % (RSD < 12.0 %) in fish tissues, and the LOQ was 5.0 μg kg−1. The results of the exposure study reveal that eugenol is eliminated within 96 h. The proposed method is effective for determining eugenol in fish, and could be used for eugenol-residue monitoring of fish.
References
Popovic NT, Strunjak-Perovic I, Coz-Rakovac R, Barisic J, Jadan M, Berakovic AP, Klobucar RS (2012) Tricaine methane-sulfonate (MS-222) application in fish anaesthesia. J Appl Ichthyol 28:553–564
Weber RA, Peleteiro JB, García Martín LO, Aldegunde M (2009) The efficacy of 2-phenoxyethanol, metomidate, clove oil and MS-222 as anaesthetic agents in the Senegalese sole (Solea senegalensis Kaup 1858). Aquaculture 288:147–150
Li JC, Liu H, Yu MQ, Wu LD, Wang Q, Lv HY, Ma B, Song Y (2014) Rapid determination of tricaine mesylate residues in fish samples using modified QuEChERS and high performance liquid chromatography-tandem mass spectrometry. Anal Methods 6(22):9124–9128
Zhang YW, Chen YW (1997) Isobiflorin, a chromone C-glucoside from cloves (Eugenia caryophyllata). Phytochemistry 45:401–403
Meinertz JR, Schreier TM, Porcher ST, Smerud JR, Gaikowski MP (2014) Depletion of eugenol residues from the skin-on fillet tissue of rainbow trout exposed to 14C-labeled eugenol. Aquaculture 430:74–78
Delbon MC, Paiva MJTR (2012) Eugenol in tilapia juvenile: concentrations and successive administrations. Bol Inst Pesca 38(1):43–52
Thompson DC, Barhoumi R, Burghardt RC (1998) Comparative toxicity of eugenol and its quinone methide metabolite in cultured liver cells using kinetic fluorescence bioassays. Toxicol Appl Pharmacol 149(1):55–63
Anderson WG, McKinley RS, Colavecchia M (1997) The use of clove oil as an anaesthetic for rainbow trout and its effects on swimming performance. North Am J Fish Manag 17:301–307
Japanese Positive List System for Agricultural Chemical Residues in Foods – Maximum Residue Limits (MRLs) List of Agricultural Chemicals in Foods. http://www.m5.ws001.squarestart.ne.jp/foundation/agrdtl.php?a_inq=51100
Pathak SB, Niranjan K, Padh H, Rajani M (2004) TLC densitometric method for the quantification of eugenol and gallic acid in clove. Chromatographia 60(3–4):241–244
Li Y, Sun Z, Zheng P (2004) Determination of vanillin, eugenol and isoeugenol by RP-HPLC. Chromatographia 60(11–12):709–713
Kildea MA, Allan GL, Kearney RE (2004) Accumulation and clearance of the anaesthetics clove oil and AQUI-STM from the edible tissue of silver perch (Bidyanus bidyanus). Aquaculture 232(1–4):265–277
Beaudry F, Guénette SA, Vachon P (2006) Determination of eugenol in rat plasma by liquid chromatography-quadrupole ion trap mass spectrometry using a simple off-line dansyl chloride derivatization reaction to enhance signal intensity. Biomed Chromatogr 20(11):1216–1222
Meinertz JR, Hess KR (2014) Evaluation of analytical techniques to determine AQUI-S®20E (eugenol) concentrations in water. 418–419:62–66
Meinertz JR, Porcher ST, Smerud JR, Gaikowski MP (2014) Determination of the exposure parameters that maximize the concentrations of the anesthetic/sedative, eugenol, in rainbow trout (Oncorhynchus mykiss) skin-on fillet tissue. Food Addit Contam 31(9):1522–1528
Chen Y, Guo Z, Wang X, Qiu C (2008) Sample preparation. J Chromatogr A 1184:191–219
Valsecchi S, Rusconi M, Polesello S (2013) Determination of perfluorinated compounds in aquatic organisms: a review. Anal Bioanal Chem 405:143–157
European Commission Decision 2002/657/EC (2002) Implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Eur Commun L221
Acknowledgments
This research was supported by the Basic Scientific Research Business Expenses Special Funds under the Chinese Academy of Fishery Sciences (2013C005).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, J., Zhang, J. & Liu, Y. Optimization of solid-phase-extraction cleanup and validation of quantitative determination of eugenol in fish samples by gas chromatography–tandem mass spectrometry. Anal Bioanal Chem 407, 6563–6568 (2015). https://doi.org/10.1007/s00216-015-8823-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8823-y