
Abstract
The development of a liquid chromatography high-resolution mass spectrometry quadrupole-time-of-flight (LC-HRMS-QTOF) method for the analysis of new stimulant designer drugs (e.g. phenethylamine, amphetamine, cathinone and piperazine derivatives) and common drugs of abuse (e.g. ketamine and ritalinic acid) in urine is reported. Sample preparation was carried out by a fast and convenient salting-out liquid-liquid extraction (SALLE) procedure. The data was generated by a preferred target list combined with untargeted data-dependent acquisition recording additional sample information (i.e. not listed metabolites of target compounds or not database-stored drugs). The identification is realised by a fully automated data extraction algorithm, taking into account accurate mass spectra, fragment masses and retention times. Method validation comprised selectivity, linearity, accuracy, stability, determination of the limit of detection (LOD) and limit of quantification (LOQ) and evaluation of matrix effects and recoveries for a total set of 39 compounds. Acceptable quantitative results were obtained for 35 of the 39 analytes. Exemplarily, application of the additional untargeted data-dependent acquisition mode enabled the identification of metabolites of the preferred target list compounds ketamine and methylenedioxypyrovalerone (MDPV) without use of reference standards. Therefore, improvement of the database is feasible with every positive library hit. The approach presented here provides a very useful tool for the combined targeted and untargeted analysis of drugs of abuse in biological matrices such as urine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, the availability of new designer drugs entering the illicit drug market has increased tremendously. Many chemical entities of those novel stimulants are derived from already known psychoactive substances, such as cannabinoids, cathinones, phenethylamines, amphetamines, tryptamines and piperazines [1–3]. These designer drugs are sold as “Spice”, “Legal Highs”, “Bath Salts”, “Plant-Growth Fertilizers” or “Research Chemicals” and are mainly distributed over the Internet. Cathinones are β-keto analogues of amphetamine-type molecules and have amphetamine-like effects due to their structural similarity [4]. Phenethylamines and tryptamines became popular as recreational drugs after the publication of two books by the American pharmacologist Alexander Shulgin [5, 6]. Piperazines, such as benzylpiperazine for example, were found in online-sold “Legal High” mixtures often in combination with other drugs (e.g. amphetamine, cocaine and ketamine) [1]. Most of the new substances have not been evaluated critically in controlled clinical studies, and nothing is known specifically about severe side effects and possible toxic metabolites. Nearly every month, there are new entities reaching the market making it almost infeasible to deal with [7–9]. In many cases, they cannot be detected in common drug screening assays where only classical stimulants (e.g. cocaine, amphetamine and methamphetamine) are monitored by highly selective methods such as targeted GC-MS, LC-MS and LC-MSMS.
In our area of Germany, North-East Bavaria (border area to the Czech Republic, a country with a more liberal drug policy), we experienced an increasing demand from hospitals and addiction clinics for the clinico-toxicological analysis of new stimulant drug types, especially concerning amphetamine-type derivatives (e.g. cathinones, piperazines and phenethylamines). Thus, we needed an analytical approach to deal with this current issue. Several approaches to solve the problem of the identification of new drug entities are published in literature: Dresen et al. reported on the identification of 700 drugs by multi-target screening with a LC-MS/MS system and library searching, containing electrospray ionisation-MS/MS spectra of over 1,250 compounds [10, 11]. Maurer et al. published unequivocally reproducible GC-MS spectra of a total of 8,650 clinically relevant substances, including 5,200 metabolites in a comprehensive GC-MS database [12]. In addition, Roesner et al. published a mass spectral library comprising over 20,000 mass spectra which cover the entire range of designer drugs up to December 2013 [13]. Concheiro et al. reported on a high-resolution (HR)-LC-MS method able to quantify 28 synthetic cathinones in urine [14]. Other promising approaches, using HR-LC-MS-quadrupole-time-of-flight (QTOF) in forensic and clinical toxicology, were applied by Bröcker et al. and Sundström et al. [15, 16]. Besides the good mass resolving power of TOF devices, one further great advantage in QTOF mass spectrometry is the data-dependent acquisition of non-targeted compounds. Hence, it is possible to record almost every generated ion in a data file and reanalyse this file in case retrospectively. After critical evaluation of the published data in literature, the QTOF approach has been the most promising in our opinion. Hence, a data-dependent and scheduled LC-HRMS-QTOF method for the simultaneous identification of 62 compounds in urine and semi-quantification of 35 compounds thereof has been developed. The here published database entries cover mainly amphetamines, phenethylamines, cathinones, piperazines and other drugs of abuse (e.g. ketamine, phencyclidine, ritalinic acid, etc.). In particular, the here presented approach, on the contrary to the work of Bröcker et al. and Sundström et al. [15, 16], combines a preferred target ion screening with a data-dependent MSMS spectra generation of even not listed targets (i.e. metabolites). Bröcker et al. merely describe the acquisition in data-dependent auto-MS mode without use of a preferred target list. Thus, the data are generated statistically depending on the abundances of any of the precursors, making targeted analysis intricate. Sundström et al. use the so-called broad-band collision-induced dissociation (bbCID; other synonym, all-ions mode), where every ion in the sample is fragmented at two collision energies (i.e. 8 and 30 eV) without single isolation of specific precursors. Even though this is also a very interesting approach, it may be susceptible to accidentally co-eluting matrix components giving identical mass fragments as the target analytes do, and hence falsifying quantitative results. In this case, a MS spectrum cannot be unambiguously traced back to its precursor ion. This could be especially severe in the realm of small molecules, where identical molecular fragments may occur more often. Therefore, the combination of a data-dependent algorithm combined with a preferred target list seemed to be more promising in our opinion.
Material and methods
Chemicals, standards and samples
Formic acid (eluent additive for LC-MS, 98 %) and ammonium formate (eluent additive for LC-MS) were obtained from Sigma-Aldrich (Steinheim, Germany). Acetonitrile (ACN) ROTISOLV® (LC-MS grade, 99.95 %) and methanol (MeOH) ROTISOLV® (LC-MS grade, 99.95 %) were purchased from Roth (Karlsruhe, Germany). HPLC-grade water was generated with an arium® 611/613 water purification system (Sartorius Stedim Biotech GmbH, Göttingen, Germany). Ammonium acetate (for analysis) and potassium hydroxide (KOH; pellets for analysis) were purchased from Merck (Darmstadt, Germany). Drug-free synthetic urine was obtained from nal von minden GmbH (Regensburg, Germany). Drug-positive and drug-negative real human urine samples were obtained from several of our customers (hospitals, addiction clinics and driver’s licence regaining institutes). Standards and internal standards, used for quantification and quality control samples (QCs), were purchased from Lipomed (Weil am Rhein, Germany) and LGC Standards (Wesel, Germany) and are shown in Table 1.
LC conditions
The LC system consisted of an Agilent Infinity 1290 SL binary pump with an integrated two-channel solvent degasser, a thermostated Agilent 1290 HiP-ALS autosampler system (20 μL injection loop) and a 1290 Agilent TCC SL column compartment (Agilent Technologies, Santa Clara, USA). Target analytes were separated with a ZORBAX Eclipse Plus C18 (100 mm × 2.1 mm I.D., 1.8 μm) column (Agilent Technologies, Waldbronn, Germany). A phenomenex SecurityGuard Ultra cartridge (C18, 10 mm × 2.1 mm I.D., < 2 μm) was used for column protection (phenomenex, Aschaffenburg, Germany). Chromatographic conditions were as follows: mobile phase A consisted of a 5 mM ammonium formate buffer, adjusted to pH 4 with formic acid. Mobile phase B consisted of pure acetonitrile with 0.1 % formic acid. The flow rate was 0.4 mL/min, and the column oven temperature was constantly kept at 40 °C. Chromatography was performed beginning with isocratic elution at 5 % B for 1.5 min, subsequently continuing into a shallow linear gradient to 35 % B in 6 min (run time step 7.5 min) and followed by a steep linear gradient to 90 % B in 0.37 min (run time step 7.87 min). The level of 90 % B was held for 1.13 min (run time step 9.00 min) in order to remove any lipophilic compound and linearly set back to 5 % B in 0.50 min (run time step 9.50 min). The isocratic period was kept at 5 % B for another 2.50 min to equilibrate the column again (total run time 12 min). The injection volume was 6 μL.
MS-QTOF conditions
The MS system consisted of an accurate 6550 iFunnel Q-TOF instrument (Agilent Technologies, Santa Clara, USA) operated with an electrospray ion source (dual-spray ESI source). A quadrupole served for isolation of precursor ions (mass window 1.3 m/z) in data-dependent acquisition mode (auto-MS), and a linear hexapole collision cell (nitrogen as collision gas) was used for precursor fragmentation. The masses of precursors and fragments were measured with the TOF-MS module giving a mass accuracy of at least <5 ppm for precursor ions and <10 ppm for product ions. TOF acquisition was operated in high-sensitivity mode and the detection frequency in dynamic range 2-GHz mode (200,000 points/transient). The mass resolving power at full-width half maximum (FWHD) was within 9,000–15,000 (m/z range 64.0158 to 322.0480, representing the sodium adduct of acetonitrile and the protonated molecule of hexakis(2,2-difluoroethoxy)phosphazine, respectively) in the high-sensitivity mode. The scan rate was adjusted to the small molecules application (<1,700 m/z). Mass calibration of the TOF system was continuously controlled by measuring the protonated reference ions of purine ([M+H]+ = 121.050873) and hexakis (1H,1H,3H-tetrafluroropropoxy)phosphazine ([M+H]+ = 922.009798). Source ionisation parameters were as follows: ESI was operated in positive ionisation mode. The nebulizer was set to 35 psi, sheath gas temperature to 380 °C, sheath gas flow to 11 L/min, drying gas temperature to 150 °C, drying gas flow to 11 L/min, capillary voltage to 3,000 V and the nozzle voltage to 0 V. The following ion funnel conditions were used: funnel DC at 30 V, funnel voltage drop at 100 V (high pressure) and 50 V (low pressure) and RF voltage at 90 V (high pressure) and 40 V (low pressure). The QTOF device was operated in data-dependent acquisition mode (auto-MS) using a preferred target ion list. Ions between 50 and 1,000 m/z were recorded in MS mode and from 40 to 500 m/z in MS/MS mode. The MS scan rate was 5 Hz and the MS/MS scan rate was 6 Hz. The maximum number of precursors per MS cycle was set to four, and priority sorting thereof was executed by abundance only mode. The minimum abundance for precursor selection was set to 3,000 counts as threshold, and every selected precursor was actively excluded for 3 s. Thus, a full cycle contained the following time segments: 200 ms scanning in MS mode (5 Hz), then a maximum of four precursors (minimum abundance of 3,000 counts) were picked for the MSMS fragmentation in auto-MS mode, of which each precursor had been scanned with a MSMS frequency of 6 Hz and thus 167 ms. This leads to the full cycle time 200 ms (5 Hz MS mode) + 4 × 166 ms (6 Hz MSMS mode for each precursor) + 100 ms (extra time needed for the acquisition readjustment) = 964 ms for one cycle, meaning that within this time, ca. 1 s, four precursors could be scanned in MSMS mode. The averaged chromatographic peak width was 6 s, approximately giving the theoretical possibility of scanning 12 different precursors co-eluting within one peak by taking into account the active exclusion of 3 s (4 precursors × 6 s = 24 spectra, theoretically giving two spectra for each compound). The isolation width of the quadrupole was adjusted to narrow (∆ = 1.3 m/z). Standardly, every precursor’s collision energy (CE) was following the linear expression CE (eV) = (m/z × 6 eV / 100) + 4 eV. Since we were mainly interested in known target compounds, a data-dependent preferred precursor table was implemented. Thus, independent of the abundance of any precursor, a preferably listed precursor was chosen for MS/MS experiments. Within the preferred list, precursors were chosen according to their abundances again. The preferred list analytes are shown in Table 1. Listed precursors (single charged) were picked for fragmentation experiments dependent on the following parameters: ∆ mass < 20 ppm and retention time ±0.3 min. MassHunter software packages (Agilent Technologies, Santa Clara, USA) were used for data acquisition (Version B.05.01), as well as qualitative analysis (Version B.06.00) and quantitative analysis (Version B.05.02). The personal compound database and library manager (PCDL, Version Rev. B.04.01, Agilent Technologies, Santa Clara, USA) was used for verification of positive compound identification results obtained by the MassHunter Qualitative Analysis software. For the evaluation and structural elucidation of not database-stored metabolites, the MassHunter Molecular Structure Correlator software tool (Version B.05.00, Agilent Technologies, Santa Clara, USA) was used. Some of the in-house PCDL database compounds are also shown in Table 1. Library spectra were obtained similar as described by Bröcker et al. [15].
Data mining algorithms
The whole database contained at that time 121 mono-isotopic mass entries. QTOF-MSMS spectra thereof were available for 81 compounds of which 62 spectra were obtained by reference standards. The remaining 19 product-ion spectra and 40 mono-isotopic mass entries were stored in the in-house Forensics-PCDL database (Bröcker, Herre & Pragst) from Agilent Technologies. Retention time data were recorded for the 62 reference standards. Exemplary compounds are presented in Table 1. Identification of target analytes was achieved by application of the combination of the Find by AutoMS (AutoMS) and Find by Formula (FbF) data extraction algorithms, which are both implemented in the MassHunter Qualitative Analysis software tool. The AutoMS algorithm matched every generated MS/MS spectrum (tolerance precursor <10 ppm; tolerance product-ion <20 ppm) with an in-house spectrum library. The FbF algorithm matched every compound contained in the in-house database (spectrum library) according to its exact mass (Δmass <10 ppm) and isotopic pattern, including again the exact masses of isotopes (M + 13C and M + 2 × 13C), the calculated abundances of all isotope masses of interest and the isotope spaces between them. Finally, the software gave scores useful for the identification of questionable compounds. Additionally, the retention time was considered. Every compound, identified by MS and MSMS, had to lie within a retention time window of ±0.1 min. Without this condition, the result was not accepted, especially if severe deviations from the expected retention area had occurred. Thus, a correct MSMS spectrum with a product-ion acceptance of normally <10 ppm and the fulfilment of the retention time criterion were absolutely mandatory, beside the correct protonated molecule mass (normally <5 ppm), for a positive identification result. The specification criteria are shown in Table 2 and further explained by a real sample result in Fig. 1. Unknown compounds (i.e. metabolites) and MSMS fragments thereof were analysed by the MassHunter Molecular Structure Correlator software tool and compared with literature results, if available.

Upper chromatogram (counts vs. acquisition time): extracted peak chromatogram of a real-case human urine sample with ISTD (mephedrone-D3), ketamine and its co-eluting metabolites norketamine and dehydronorketamine (rhombuses indicate where a MSMS spectrum was taken). Left spectrum (counts vs. mass-to-charge): MS precursor scan (above) and MSMS product scan (below) of ketamine. Right spectrum (counts vs. mass-to-charge): extracted isotopic peak pattern (by FbF algorithm) of ketamine. Table above: the identified compound (ketamine) with its appropriate scores (FbF and AutoMS), mass difference in ppm, retention time (RT) and retention time difference (RT Diff) and peak area. Table below: isotope scores for mass accuracy (Score (mass)), isotopic abundance (Score (iso. abund.)) and isotopic spacing (Score (iso. spacing)). According to the I.D. criteria (see Table 2), the compound (ketamine) has obtained 4 out of 4.5 I.D. points, if the score (mass) with 89.72 is evaluated strictly
Reagents and standards preparation
The salting out reagent (10 M ammonium acetate) was prepared by dissolving 77.08 g of ammonium acetate in 100 mL of LC-MS-grade water. A potassium hydroxide solution (ca. 10 M) was prepared by dissolving 56.11 g of KOH pellets in 100 mL of LC-MS-grade water. Different concentrations of standard stock solutions (1, 0.5, 0.4 and 0.1 mg/L) in methanol (MeOH), containing the substances shown in Table 1 (39 compounds and 7 internal standard (ISTD)), were used for sample preparation. Corresponding dilutions in the range of 25–250 μg/L (for validation procedures assessing the linearity, six-point calibration with 25, 50, 100, 150, 200 and 250 μg/L) and 25–100 μg/L (for daily routine operations, two-point calibration with 25 and 100 μg/L) were prepared in drug-free human urine (for validation procedures) or in synthetic urine (for daily routine operations). Dilution ranges for the evaluation of the limit of quantification (LOQ) and limit of detection (LOD) were 2.5–25 μg/L (six-point calibration) and were evaluated in drug-free human urine. The internal standard stock solution (0.5 mg/L) contained deuterated analogues of the target compounds as listed in Table 1. Quality control samples (QCs) at two different levels (QClow with 30 μg/L and QChigh with 60 μg/L) for the determination of accuracy, precision and the matrix effect were prepared in drug-free human urine (for validation procedures) and in synthetic urine (for daily routine operations).
Sample preparation
The extraction procedure is based on a salting-out liquid-liquid extraction (SALLE) approach, similarly related to the descriptions given in the publication of Yanes and Lovett [17]. To a volume of 200 μL blank urine in a 1.5-mL microcentrifuge tube, spiked with drugs at the appropriate concentration level, a volume of 20 μL internal standard (c = 0.5 mg/L) was added. Then, 250 μL of acetonitrile was added and the tube was mixed by vortexing for 5 s. An aliquot of 10 M ammonium acetate solution (50 μL), followed by an aliquot of 10 M KOH solution (50 μL), was added successively, and the mixture was vortexed for 5–10 s. Afterwards, the microcentrifuge tubes were centrifuged at 10,000 rounds per minute (rpm) for 3 min. From the upper organic layer, 100 μL was removed and transferred to a LC vial. In order to adjust the polarity of the analyte solution to the starting LC conditions, 100 μL of mobile phase A was added and the final diluted extract was mixed by vortexing for 5 s again. The injection volume for the chromatographic device was 6 μL.
Validation parameters
Validation of the method presented here included selectivity, linearity, precision, accuracy, matrix effects, extraction recovery and determination of the LOD and LOQ, respectively. Processed sample stability was assessed during the determination of linearity (total work list run times greater than 24 h). Three freeze/thaw cycles were evaluated in drug-free human urine at concentration levels of 25 and 50 μg/L. Further comprehensive stability studies (i.e. long-term stability) were not explicitly evaluated as they are already published in literature for most of the analytes discussed here [18–21, 14]. The Microsoft Excel®-based software package Valistat 2.0 (ARVECON GmbH, Walldorf, Germany) was used for calculations. All quantification results are based on MS traces of the TOF device. MS/MS spectra are solely used for compound identification. The focus of the method has been the unambiguous qualitative assignment of drugs of abuse. Since only an approximate estimation of concentrations has been needed by our customers, the method validation concerning quantitative results was reduced to the least necessary evaluations. This comprised, for example, merely a two-point calibration in routine analysis and assessment of matrix effects with only six drug-free negative human urine samples at two levels, giving for each level three different processed specimens instead of five as usually claimed by bioanalytical validation protocols [22]. Therefore, the whole assay has been stated from the beginning as a semi-quantitative approach.
For evaluation of selectivity, 20 drug-free human urine samples were prepared according to the SALLE extraction protocol explained before in the section “Sample preparation”. The generated data-dependent raw data files were then analysed by the AutoMS and FbF data extraction algorithms of the MassHunter Qualitative Analysis software tool (see also the section “Data mining algorithms”).
For determination of linearity, six aliquots of drug-free human urine, each spiked with standard at the appropriate level, were analysed (calibration levels 25, 50, 100, 150, 200 and 250 μg/L; ISTD concentration 50 μg/L). Each calibration level was injected six times in order to evaluate the precision of the LC-QTOF system. The obtained peak area ratios (analyte/ISTD) were plotted against the corresponding drug concentrations of each analyte.
The LOD and LOQ were evaluated by injection of another six drug-free human urine aliquots, each spiked with standard at the appropriate level (calibration levels 2.5, 5, 10, 15, 20, 25 μg/L; ISTD concentration 50 μg/L). Each calibration level was injected three times, and the obtained peak area ratios (analyte/ISTD) were plotted against the drug concentrations of each analyte. The procedure for evaluation of the LOD and the LOQ, respectively, followed the recommendations of the German DIN standard 32645 [23]. Additionally, the signal-to-noise ratio (S/N) was considered for some compounds (LOD with S/N = 3; LOQ with S/N = 10).
The accuracy, intra- (RP) and inter-day (LP) imprecision data were obtained by evaluation of two quality control samples (QCs) at low and high concentration levels (QClow with 30 μg/L and QChigh with 60 μg/L). Determination of the accuracy data was realised by a two-point calibration (calibration levels 25 and 100 μg/L) as considered more practically for daily routine operations. It should be mentioned that the QCs were not pooled and prepared daily from cooled QC standards (−20 °C in MeOH) in drug-free urine and synthetic urine, respectively. QCs were evaluated at least for 8 days (in duplicate) for every analyte.
Matrix effects and extraction recovery were studied according to the protocol published by Matuszewski et al. [24] and Yanes and Lovett [17]. This approach, according to the SALLE protocol described by Yanes and Lovett [17], is based on the following theoretical assumptions: The analytes in 200 μL of urine are completely extracted into 250 μL of acetonitrile (dilution factor 0.8) after the addition of 10 M ammonium acetate (50 μL) and 10 M KOH (50 μL) and phase separation after centrifugation. Besides, 100 μL from the 250 μL of acetonitrile is further diluted with 100 μL of mobile phase buffer A (dilution factor 0.4). Thus, only 40 % of analyte of the 200 μL urine sample is recovered. This theoretical assumption is taken into account in the following explanations. The approach contains three sets: set A (sample without matrix), set B (drug-free urine extracted and drug added after extraction) and set C (extraction of urine with drugs added before extraction). Set A gives the absolute signal intensity without any matrix and extraction interferences. Set B reveals the influence of the matrix on the signal intensity. Set C shows the influence of the extraction and matrix on the signal intensity. Each set was evaluated at two levels (30 μg/L for QClow and 60 μg/L for QChigh). At each level, three different drug-free human urine samples were used for the evaluation of matrix effects (set B) and extraction recovery (set C). Thus, a total number of 18 runs were given (six for QClow/high in set A, six for QClow/high in set B and another six in set C for QClow/high). Set A: A volume of 200 μL QC solution, diluted in LC mobile phase buffer A (5 mM ammonium formate adjusted to pH 4 with formic acid), at the appropriate concentrations (30 and 60 μg/L) was further diluted with 50 μL LC buffer A (in order to simulate the dilution factor of 0.8), and 20 μL ISTD (c = 0.5 mg/L) was added. From this solution, 100 μL was further diluted with 100 μL of acetonitrile in a HPLC vial giving the absolute signal intensity without any matrix and extraction interferences. Set B: A volume of 200 μL drug-free human urine was further diluted with 20 μL of MeOH (to adjust the polarity similar to set C, where the ISTD is given at this step), and 250 μL acetonitrile was added and shortly mixed by vortexing. Then, 50 μL of ammonium acetate (10 M) and 50 μL of KOH (10 M) were added successively, and the mixture was again vortexed and centrifuged (10,000 rpm for 3 min). From the upper organic layer, 90 μL was added into a HPLC vial and 10 μL of ISTD (c = 0.4 mg/L) was given (in order to correctly simulate the dilution factor for the ISTD signal). A volume of 100 μL appropriate QC solution (30 and 60 μg/L, diluted with factor 0.8, to simulate the extraction with 250 μL ACN, giving concentrations at 24 and 48 μg/L, respectively), diluted in LC mobile phase buffer A (similar to set A), was added and mixed by vortexing. Set C: The samples, 200 μL of drug-free human urine, spiked with drugs at the appropriate QC level (30 and 60 μg/L), were prepared as described in the section “Sample preparation”. Dividing set B through set A reveals the influence of the matrix on the signal intensity, and dividing set C through set B leads to the extraction recovery. The overall process efficiency can be obtained by dividing set C through set A.
Results and discussion
Method validation
The here reported method showed a very good selectivity due to the efficient mass resolving power of the time-of-flight device. The analysis of 20 drug-free human urine samples did not reveal any false-positive result when, as explained before in the section “Data mining algorithms”, MSMS fragments and the retention time were considered as the crucial identification criteria. Critical evaluation of the data revealed that if only protonated molecule masses are considered, there would be a certain amount of false positives (data not explicitly shown here) due to the relatively wide delta mass range (±10 ppm was extracted) used for data extraction. Thus, a good match of a MSMS spectrum is still mandatory.
All analytes proved to be linear within a certain range as shown in Table 3. In some cases, one of the calibration levels was eliminated leading to differing linearity ranges. Calibration curves of all target compounds were generally acceptable (correlation coefficients R 2 > 0.98). In particular, standards with their deuterated analogues (e.g. methylone, amphetamine, MDA, MDMA and PCP) usually revealed better results than standards without. As discussed before, no severe interfering of matrix with compounds of interest was observed. Evaluation of the injection precision of the system was acceptable, and no significant outliers were detected. However, the two simplest cathinone derivatives, cathinone and its N-methylated analogue methcathinone, were only linear up to 150 μg/L. Concentrations at 200 and 250 μg/L, respectively, exhibited a saturation phenomenon or, as in the case for cathinone, already started to decompose. Since it is known in literature that cathinone derivatives tend to degrade in solution at high pH values and temperatures [20, 19, 21, 14], the autosampler temperature was thermostated at 10 °C. Nevertheless, cathinone never showed an acceptable processed sample stability and is anyway primarily metabolised to cathine (norpseudoephedrine and norephedrine, respectively) and hence may not be detectable in urinary specimens [25]. The other cathinones (e.g. methylone, mephedrone, methedrone, butylone, 4-methyl-N-ethylcathinone (4-MEC) and methylenedioxypyrovalerone (MDPV)) were stable under the experimental conditions. Basically, all analytes, except cathinone, were stable within 24 h in the autosampler at 10 °C. Methcathinone seems to be more stable than cathinone, but may just decompose more slowly and is also preferably metabolised to pseudoephedrine and ephedrine [25].
We obtained acceptable values for the LOD (<10 μg/L) and LOQ (<25 μg/L). As we used the German DIN standard 32645 method [23] for evaluation of the LOD and LOQ, the values were greater than the classic approach (LOD = S/N × 3 and LOQ = S/N = × 10) would give in some cases. By evaluation of the signal-to-noise ratio instead, some of the analytes would get easily below 1 μg/L as LOD. However, since we focused on high selectivity by the MS (5 spectra/s) and MSMS data acquisition algorithm (6 spectra/s) used, some transients are lost and the signal strength diminishes at the cost of LOD and LOQ, respectively. But, in order to generate enough MSMS spectra for all 47 compounds (standards and ISTDs) co-eluting in the QCs, this speed was necessary and generally generates more MSMS spectra of even other compounds probably occurring in the sample (i.e. metabolites not listed in the preferred list). Another aspect has been that in daily routine, where only a two-point calibration is applied (25 and 100 μg/L), every positive hit with a concentration value below 25 μg/L would be communicated as identified below the calibration range (<25 μg/L). In questionable cases (signal too low), it is anyway possible to do a second analysis where a compound of interest is specifically monitored. This could be achieved manually (i.e. up-scale: more sample is extracted, 500 μL instead of 200 μL of urine) or digitally by focusing on only a few specific ions and using another scan rate (i.e. MS at 2 spectra/s and MSMS at 3 spectra/s, which would mean an increase of approximately factor 2 for the signal intensity).
The accuracy results were all obtained by a two-point calibration approach. From the total of 39 compounds, only 20 did reveal relatively robust accuracy results (imprecision RSD <15 % and bias <15 % [22]). A few compounds (e.g. mCPP, MDPV or 2C-T-4, each with a RSD slightly higher than 15 %) had greater deviations, at least for one QC level (high or low). Other compounds (e.g. 3,4-DMA; PMA; 2C-H; 3,4,5-TMA; BDB; 4-MTA, methylphenidate, 2C-B and DOB) did show stronger deviations, especially concerning the inter-day imprecision results. The QC accuracy levels revealed in almost all samples a systematic bias below the theoretical concentrations. Reasons therefore may be the low sample volumes worked with (200 μL urine), the daily preparation of QCs (no QC pools were built), the relatively low QC levels (30 and 60 μg/L) checked and, additionally, the missing adequate internal standard references for the majority of compounds. Standards with an internal standard reference (e.g. methylone, amphetamine, methamphetamine, MDA, MDMA, mephedrone and PCP) generally had a better precision and accuracy. In cases where deviations were too strong (e.g. cathinone, methylphenidate, PMA and 4-MTA) and thus not really reproducible, the analysis thereof is merely performed qualitatively without any quantitative result hitherto. However, all accuracy results (RP, LP and bias) lay within an interval of at least 75 to 125 %, except for PMA, methylphenidate and 4-MTA. Therefore, the method has to be stated as a semi-quantitative approach and may be only applied qualitatively in questionable cases.
Matrix effects approximately ranged from 50 to 300 %. Extremely strong ion enhancing effects were detected for 3,4-DMA (324 % at QClow) and ritalinic acid (293 % at QClow) and also for cathinone (165 % at QClow) and 3,4,5-TMA (163 % at QClow). Even after repetition of the matrix effect testing (second set with 18 runs), these four analytes always revealed a powerful ion enhancement. Nevertheless, quantitative determinations for them (3,4-DMA; 3,4,5-TMA and ritalinic acid) have been still reproducible. The other compounds showed moderate matrix effects, and at the end of the chromatographic gradient, almost no real matrix influence is apparent. Compared to the QCs, the real-case samples can be regarded anyway as less prone to such matrix effects as found in QCs since they are overloaded with co-eluting drug material. A real-case sample probably will not reveal as many drugs as detectable in the QCs. On average, the recovery of almost any compound was constantly around 50 %, which means that by the here applied extraction approach (SALLE) only 50 % of the original sample amount has been transferred into the organic phase. Merely ritalinic acid gave a weaker recovery result (QClow 23 % and QChigh 31 % on average) which is due to its amphoteric chemical character. However, compared with commonly known urine dilution methods (i.e. 1:10 with water) the SALLE approach definitely leads to purer extracts. Another advantage, though at the cost of recovery, is the fast and convenient sample preparation achieved by the SALLE approach.
As already discussed, all analytes, except for cathinone, were stable in the autosampler at 10 °C within 24 h (determined by repetitive measurements of the QCs at 30 and 60 μg/L; see also Table 3 repeatability imprecision RP). Cathinone was only stable up to 5 h (evaluated during the assessment of linearity). Freeze/thaw stability at 25 and 50 μg/L was acceptable for all analytes, except for cathinone again. Thus, after three consecutive freeze/thaw cycles (frozen for 24 h and thawed for at least 1 h), almost all analytes from Table 3 have been reproducibly detectable (within the respective inter-day imprecision LP intervals), not showing any significant decrease in their concentrations. This has been especially surprising for some cathinones (methcathinone, methylone, mephedrone, methedrone, butylone, 4-MEC and MDPV) since we expected at least a minor decrease in amounts for some of them. Anyhow, due to the relatively high inter-day imprecision (LP) of the method for some compounds (SD > 15 %), a minor decrease of any of the analytes cannot be absolutely excluded yet. For further details, see Table 3.
Practical application
In order to demonstrate the applicability of the here presented procedure, two examples of real cases are presented in which it has been successfully applied.
Ketamine and metabolites
In two human urine samples, out of a series of 76 screened samples, two were positive for ketamine and its metabolites dehydronorketamine and norketamine (positive rate of 2.6 %). Quantitative values were 14 and 21 μg/L, respectively, and thus, the results have been stated as identified with <25 μg/L. The overview data is already presented in Fig. 1. As can be seen there, even without possession of the corresponding metabolite standards from ketamine (norketamine and dehydronorketamine), the data-dependent method is able to additionally deliver data thereof. Every compound was correctly assigned by the FbF algorithm which elucidated the remarkable isotopic pattern due to the presence of chlorine (isotopic distribution of chlorine-35 and chlorine-37 with an abundance of approximately 3 to 1). Furthermore, the MSMS spectra of both norketamine and dehydronorketamine were recorded though they were not listed in the preferred table of the data acquisition tool. The MS and MSMS spectrum of dehydronorketamine is depicted exemplarily in Fig. 2. Thus, we were able to ensure the positive identification of ketamine and its metabolites in urine even without the use of expensive reference standards. The here obtained data of norketamine and dehydronorketamine (MSMS spectra and retention time) have been added to our database. Comparison with MSMS results from literature verified their identity unequivocally [26, 27]. Starting from m/z 238.1006 (Δ = −3.1 ppm) for ketamine, the loss of methylamine (CH3NH2) led to fragment m/z 207.0585 (Δ = 4.0 ppm), followed by a loss of CO which gave the m/z 179.0608 (Δ = 10.9 ppm). A loss of water (H2O) led to the fragment m/z 220.0887 (Δ = 2.7 ppm), and fragmentation of methylamine gave m/z 189.0465 (Δ = 3.2 ppm). The base peak at m/z 125.0152 (Δ = 4.8 ppm) may be explained by a neutral loss from m/z 179 and m/z 189, respectively. Norketamine gave similar transitions as ketamine did: m/z 224 to m/z 207 (loss of NH3), m/z 207 to m/z 189 (loss of H2O), m/z 207 to m/z 179 (loss of CO) and from m/z 179 and m/z 189 to m/z 125 (the 2-chloro-benzyl cation) by appropriate neutral losses. Dehydronorketamine gave the following specific fragments: Starting from m/z 222, a loss of NH3 gave the fragment m/z 205 (ca. 50 % intensity). The fragment at m/z 177 (ca. 85 % intensity) can be explained by a neutral loss of CO from m/z 205. A further loss of HCl from m/z 177 gave the signal at m/z 141 (ca. 55 % intensity). And, interestingly, the fragment at m/z 142.0772 (Δ = −3.5 ppm, C11H10) may be only explainable through loss of a chlorine radical (exact mass = 34.9688 g/mol) from m/z 177.0476 (Δ = −2.9 ppm) leading to an odd-electron species (radical cation) normally not expected in ESI-MS (additionally verified by the Masshunter Molecular Structure Correlator tool). However, this assumption may not be unlikely as there are several reports in literature where similar observations have been made (i.e. odd-electron species in the fragmentation patterns of benzodiazepines [28]). The proposed fragmentation scheme of dehydronorketamine is also presented in Fig. 2.

Proposed fragmentation pathway for the spectrum of dehydronorketamine. The base peak at m/z 142.0772 (Δ = −3.5 ppm) may be explainable by the elimination of a chlorine radical from m/z 177.0469. The existence of this unusual radical cation in ESI-MS could be then further explained by the stability of the high conjugation of the system. For further details, see text
Detection of ritalinic acid, MDPV and metabolites
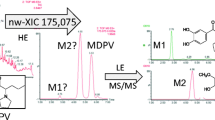
In this example, we detected in a human urine sample the following drugs: methylphenidate and its metabolite ritalinic acid and the research chemical MDPV. Methyphenidate showed a minor peak signal which was positively identified, whereas for ritalinic acid and MDPV, the observed concentrations exceeded their linearity ranges by far, leading to detector saturation of the TOF device. Thus, the extract had to be diluted (1:30) with mobile phase buffer A. Corrections by appropriate calculations gave for ritalinic acid ca. 1,800 μg/L and for MDPV ca. 350 μg/L. Both semi-quantitative results have been stated as >100 μg/L. Methylphenidate, as expected, was not detectable anymore at this dilution level. We also detected the two MDPV isomeric metabolites methylcatechol-pyrovalerone (methylcatechol-PV) already reported on by Meyer et al. and Strano-Rossi et al. [29, 30]. Furthermore, a hydrogenated metabolite of MDPV, dihydro-MDPV, was detected which has not been described in detail in literature before. The structures of the MDPV metabolites are shown in Fig. 3. In Fig. 4, the extracted compound chromatogram is depicted. The combined MS and MSMS spectra of MDPV and the two methylcatechol-PV metabolites in Fig. 4 are similar to the spectra published by Meyer et al. [29]. Hence, they may not be discussed explicitly here. More interesting has been the MSMS spectrum of dihydro-MDPV. Its compound chromatogram revealed a huge peak signal shortly eluting after MDPV (see Fig. 4). We noticed this unexpected signal after extraction of the MS trace at m/z 278.1751. At first, we thought this may be one of the methylcatechol-PV metabolites, but the mass spectrum did not match the criteria given by Meyer et al. [29]. Nevertheless, it did match, according to its retention time (shortly eluting after MDPV) and in-source fragmentation pattern (similar fragments as in Fig. 4 for dihydro-MDPV), the data published by Strano-Rossi et al. [30]. However, Strano-Rossi et al. regarded it as a methylcatechol-PV metabolite as they merely recorded the protonated molecule in high-resolution MS mode (QTOF) without any fragmentation. On the contrary, since the MSMS spectra at 4.6 and 5.1 min (Fig. 4) match exactly the high-resolution spectra of Meyer et al. [29], the compound at ca. 6.3 min, revealing the same protonated molecule mass (m/z 278.1751) as the methylcatechol-PVs do, may be the hydrogenated hydroxyl metabolite of MDPV. A possible fragmentation pathway for dihydro-MDPV is given in Fig. 5. As depicted there, starting from m/z 278.1760 (Δ = 3.2 ppm), a loss of water led to the base peak ion at m/z 260.1676 (Δ = −9.8 ppm; saturated signal, indicated by an * in Fig. 4, led to an inaccurate mass resolving power in this case). From m/z 260, the ion at m/z 191.1075 (Δ = −1.5 ppm) may be explainable through a hydride shift (from the alpha position in the pyrrolidine-ring system) leading to elimination of the cyclic imine structure. A subsequent loss of methanol (m/z 32.0262) may have led to the fragment at m/z 159.0820 (C10H11O; Δ = 6.36 ppm). The fragment at m/z 135.0453 (Δ = −8.9 ppm) may be the product of the loss of butene (from m/z 191). Interestingly, the ion at m/z 218.1176 would have been expected by elimination of propene (even-electron fragmentation). Instead, the odd-electron ion species at m/z 217.1116 (Δ = −8.8 ppm) was detected, concluding that a propyl radical had been eliminated. Further repetitions of this experiment always revealed the calculated mass at m/z 217.1097 (m/z values of 217.1095 with Δ = 1.07 ppm and 217.1103 with Δ = −2.6 ppm were obtained). Theoretical calculations (MassHunter Qualitative Analysis software) comparing the odd with the even-electron species have been always preferring the molecular formula C13H15NO2 and thus the odd-electron species. The best theoretical result for the even-electron species would be obtained by the molecular formula C10H18ClN2O (range from Δ = −0.38 to 3.32 ppm). However, since no typical chlorine isotopic pattern was observable, neither in the precursor ion nor in the product-ion spectrum, this molecular structure had been excluded. Same conclusions are given by the Molecular Structure Correlator software tool which explained the MSMS spectrum by equal fragments as described in Fig. 5. Further evidence can be found in the work of Strano-Rossi et al. where the high-resolution TOF-MS spectrum of methylcatechol-PV is depicted [30]. Evidently, the here discussed specific fragments of dihydro-MDPV (m/z 260 and especially the odd-electron species m/z at 217) are also observable in their work (m/z 260.1644 and m/z 217.1102, probably caused by in-source fragmentation). Conclusively, they also may have reported about dihydro-MDPV and not methylcatechol-PV. Even in this example, the positive identification of metabolites from MDPV was possible without any reference standard. The methylcatechol-PV metabolites have been identified unambiguously, and the existence of a dihydro-MDPV metabolite seems to be very likely. Additionally, the here obtained data of the MDPV metabolites have been added to our in-house database.

The structures of MDPV and its metabolites (from left to right): methylcatechol-PVs, dihydro-MDPV and MDPV. Interestingly, all MDPV metabolites have an exact mass of 278.1751 g/mol. For further details, see text

Upper chromatogram (counts vs. acquisition time): extracted peak chromatogram of a real-case human urine sample (diluted 1:30) with ritalinic acid (dark rhombuses indicate where a MSMS spectrum was taken), the MDPV metabolites (methylcatechol-PV isomers and dihydro-MDPV) and MDPV itself. Spectra in the middle (counts vs. mass-to-charge): MS precursor scan (above) and MSMS product scan (below) of the two methylcatechol-PV metabolites eluting at 4.6 and 5.1 min. Spectra below (counts vs. mass-to-charge): MS precursor scan (above) and MSMS product scan (below) of dihydro-MDPV (left) and MDPV (right). For further details, see text

The proposed fragmentation pathway of dihydro-MDPV. Surprisingly, the expected even-electron species at m/z 218.1176 (neutral loss of propene from the base peak at m/z 260.1645) was not observed. Merely the ion at m/z 217.1097 was observable, concluding that an odd-electron (radical cation), through loss of a propyl radical, had been created. Due to the many possible mesomeric structures, this may be the only reasonable explanation of the ion at m/z 217.1097. For further details, see text
Conclusions
We reported on a fully validated LC-MS-QTOF method for the analysis of new designer drugs and common drugs of abuse in urine, qualitatively and semi-quantitatively. The validation comprised 39 target compounds, mainly phenethylamines, amphetamines, cathinones and piperazines, as well as other common substances of abuse (e.g. ketamine, methylphenidate and its metabolite ritalinic acid). A fast and convenient sample extraction approach (salting-out liquid-liquid extraction, SALLE) was used. Method validation included determination of selectivity, linearity, accuracy, matrix effects, recovery, stability and limit of detection (LOD) and limit of quantification (LOQ). Most of the analytes were validated successfully, showing acceptable matrix effects, recoveries (ca. 50 % for each analyte on average) and accuracy results (inter-day imprecision <25 % and bias <25 %). A few did not meet the strict requirements (e.g. cathinone, methylphenidate, PMA and 4-MTA), and thus, quantitative results have to be stated with the uttermost carefulness. However, every compound can be still identified unambiguously by correct assignment of the accurate molecule mass, the presence of accurate MSMS fragments and the proper retention time. Moreover, a data-dependent acquisition mode has been included, which is able to additionally generate MS and MS/MS spectra of other possible drugs and metabolites not stored in the database yet. This approach unequivocally enabled the positive identification of metabolites from ketamine and MDPV without use of reference standards. Furthermore, it has been possible, by high-resolution MSMS spectra, to deduce the possible existence of two odd-electron species fragments from dehydronorketamine and a MDPV metabolite. Though odd-electron species are rather unusual in ESI-MS, their existence has been thoroughly reported in literature. The method has been successfully implemented into our daily routine laboratory work and is at the moment mainly used for clinico-toxicological issues requested by hospitals and addiction clinics in the area of Northern Bavaria. The here demonstrated LC-MS-QTOF technique, using a scheduled target search algorithm combined with data-dependent acquisition mode, seems to be a very promising attempt for the monitoring of the vast amount of new designer drugs and drugs in general which a toxicological laboratory encounters nowadays.
References
Zuba D (2012) Identification of cathinones and other active components of ‘legal highs’ by mass spectrometric methods. Trends Anal Chem 32:15–30
Dresen S, Ferreiros N, Putz M, Westphal F, Zimmermann R, Auwarter V (2010) Monitoring of herbal mixtures potentially containing synthetic cannabinoids as psychoactive compounds. J Mass Spectrom 45(10):1186–1194. doi:10.1002/jms.1811
Katagi M, Kamata T, Zaitsu K, Shima N, Kamata H, Nakanishi K, Nishioka H, Miki A, Tsuchihashi H (2010) Metabolism and toxicologic analysis of tryptamine-derived drugs of abuse. Ther Drug Monit 32(3):328–331. doi:10.1097/FTD.0b013e3181dcb40c
Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME (2013) Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol 168(2):458–470. doi:10.1111/j.1476-5381.2012.02145.x
Shulgin A (1991) PiHKAL—a chemical love story. Transform Press, Berkeley, California
Shulgin A (1997) TiHKAL—the continuation. Transform Press, Berkeley, California
Zaitsu K, Katagi M, Kamata H, Nakanishi K, Shima N, Kamata T, Nishioka H, Miki A, Tatsuno M, Tsuchihashi H (2010) Simultaneous analysis of six novel hallucinogenic (tetrahydobenzodifuranyl)aminoalkanes (FLYs) and (benzodifuranyl)aminoalkanes (DragonFLYs) by GC-MS, LC-MS and LC-MS-MS. Forensic Toxicol 28:9–18
Uchiyama N, Matsuda S, Kawamura M, Kikura-Hanajiri R, Goda Y (2013) Identification of two new-type designer drugs, piperazine derivative MT-45 (I-C6) and synthetic peptide Noopept (GVS-111), with synthetic cannabinoid A-834735, cathinone derivative 4-methoxy-alpha-PVP, and phenethylamine derivative 4-methylbuphedrine from illegal products. Forensic Toxicol online first: 3rd June 2013
Uchiyama N, Shimokawa Y, Matsuda S, Kawamura M, Kikura-Hanajiri R, Goda Y (2013) Two new synthetic cannabinoids, AM 2201 benzimidazole analog (FUBIMINA) and (4-methylpiperazin-1-yl)(1-pentyl-1H-indol-3-yl)methanone (MEPIRAPIM), and three phenethylamine derivative, 25H-NBOMe 3,4,5-trimethoxybenzyl analog, 25B-NBOMe, and C-N-NBOMe, identified in illegal products. Forensic Toxicol online first: 5th November 2013
Dresen S, Ferreiros N, Gnann H, Zimmermann R, Weinmann W (2010) Detection and identification of 700 drugs by multi-target screening with a 3200 Q TRAP LC-MS/MS system and library searching. Anal Bioanal Chem 396(7):2425–2434. doi:10.1007/s00216-010-3485-2
Dresen S, Gergov M, Politi L, Halter C, Weinmann W (2009) ESI-MS/MS library of 1,253 compounds for application in forensic and clinical toxicology. Anal Bioanal Chem 395(8):2521–2526. doi:10.1007/s00216-009-3084-2
Maurer HH, Pfleger K, Weber A (2011) Mass spectral and GC data of drugs, poisons, pesticides, pollutants and their metabolites, vol 4th Edition edn. Wiley-VCH, Weinheim, Germany
Roesner P, Junge T, Westphal F, Fritschi G (2013) Mass spectra of designer drugs 2014. Wiley-VCH, Weinheim, Germany
Concheiro M, Anizan S, Ellefsen K, Huestis MA (2013) Simultaneous quantification of 28 synthetic cathinones and metabolites in urine by liquid chromatography-high resolution mass spectrometry. Anal Bioanal Chem 405(29):9437–9448. doi:10.1007/s00216-013-7386-z
Broecker S, Herre S, Wust B, Zweigenbaum J, Pragst F (2011) Development and practical application of a library of CID accurate mass spectra of more than 2,500 toxic compounds for systematic toxicological analysis by LC-QTOF-MS with data-dependent acquisition. Anal Bioanal Chem 400(1):101–117. doi:10.1007/s00216-010-4450-9
Sundstrom M, Pelander A, Angerer V, Hutter M, Kneisel S, Ojanpera I (2013) A high-sensitivity ultra-high performance liquid chromatography/high-resolution time-of-flight mass spectrometry (UHPLC-HR-TOFMS) method for screening synthetic cannabinoids and other drugs of abuse in urine. Anal Bioanal Chem 405(26):8463–8474. doi:10.1007/s00216-013-7272-8
Yanes EG, Lovett DP (2012) High-throughput bioanalytical method for analysis of synthetic cannabinoid metabolites in urine using salting-out sample preparation and LC-MS/MS. J Chromatogr B: Analyt Technol Biomed Life Sci 909:42–50. doi:10.1016/j.jchromb.2012.10.013
Peters FT (2007) Stability of analytes in biosamples—an important issue in clinical and forensic toxicology? Anal Bioanal Chem 388(7):1505–1519. doi:10.1007/s00216-007-1267-2
Tsujikawa K, Mikuma T, Kuwayama K, Miyaguchi H, Kanamori T, Iwata YT, Inoue H (2012) Degradation pathways of 4-methylmethcathinone in alkaline solution and stability of methcathinone analogs in various pH solutions. Forensic Sci Int 220(1–3):103–110. doi:10.1016/j.forsciint.2012.02.005
Sorensen LK (2011) Determination of cathinones and related ephedrines in forensic whole-blood samples by liquid-chromatography-electrospray tandem mass spectrometry. J Chromatogr B: Analyt Technol Biomed Life Sci 879(11–12):727–736. doi:10.1016/j.jchromb.2011.02.010
Johnson RD, Botch-Jones SR (2013) The stability of four designer drugs: MDPV, mephedrone, BZP and TFMPP in three biological matrices under various storage conditions. J Anal Toxicol 37(2):51–55. doi:10.1093/jat/bks138
Shah VP, Midha KK, Findlay JW, Hill HM, Hulse JD, McGilveray IJ, McKay G, Miller KJ, Patnaik RN, Powell ML, Tonelli A, Viswanathan CT, Yacobi A (2000) Bioanalytical method validation—a revisit with a decade of progress. Pharm Res 17(12):1551–1557
Peters FT, Hartung M, Herbold M, Schmitt G, Daldrup T, Musshoff F (2009) Anhang B zur Richtlinie der GTFCh zur Qualitätssicherung bei forensisch-toxikologischen Untersuchungen Anforderungen an die Validierung von Analysenmethoden. ToxichemKrimtech 76(3):185–208
Matuszewski BK, Constanzer ML, Chavez-Eng CM (2003) Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem 75(13):3019–3030
Baselt RC (2011) Disposition of toxic drugs and chemicals in man, 9th edn. Biomedical Publications, Seal Beach, California
Wang KC, Shih TS, Cheng SG (2005) Use of SPE and LC/TIS/MS/MS for rapid detection and quantitation of ketamine and its metabolite, norketamine, in urine. Forensic Sci Int 147(1):81–88. doi:10.1016/j.forsciint.2004.03.031
Bijlsma L, Sancho JV, Hernandez F, Niessen WM (2011) Fragmentation pathways of drugs of abuse and their metabolites based on QTOF MS/MS and MS(E) accurate-mass spectra. J Mass Spectrom 46(9):865–875. doi:10.1002/jms.1963
Niessen WM (2011) Fragmentation of toxicologically relevant drugs in positive-ion liquid chromatography-tandem mass spectrometry. Mass Spectrom Rev 30(4):626–663. doi:10.1002/mas.20332
Meyer MR, Du P, Schuster F, Maurer HH (2010) Studies on the metabolism of the alpha-pyrrolidinophenone designer drug methylenedioxy-pyrovalerone (MDPV) in rat and human urine and human liver microsomes using GC-MS and LC-high-resolution MS and its detectability in urine by GC-MS. J Mass Spectrom 45(12):1426–1442. doi:10.1002/jms.1859
Strano-Rossi S, Cadwallader AB, de la Torre X, Botre F (2010) Toxicological determination and in vitro metabolism of the designer drug methylenedioxypyrovalerone (MDPV) by gas chromatography/mass spectrometry and liquid chromatography/quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 24(18):2706–2714. doi:10.1002/rcm.4692
Acknowledgments
The authors gratefully acknowledge Mr. Sebastian Bröcker for his technical assistance and support. Michael Paul thanks Mrs. Heike Franke (Rudolf-Böhm-Institut für Pharmakologie, Leipzig) and Mrs. Adelgunde Gräfe (Institut für Rechtsmedizin, Leipzig) from the University of Leipzig, Germany, for accepting this work as final thesis in the postgraduate programme in Toxicology and Environmental Protection.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Paul, M., Ippisch, J., Herrmann, C. et al. Analysis of new designer drugs and common drugs of abuse in urine by a combined targeted and untargeted LC-HR-QTOFMS approach. Anal Bioanal Chem 406, 4425–4441 (2014). https://doi.org/10.1007/s00216-014-7825-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-7825-5