
Abstract
An analytical method coupling online solid-phase extraction (SPE) and liquid chromatography–tandem mass spectrometry (LC-MS/MS) was developed to quantify 16 endogenous nucleoside mono- and triphosphates in cellular samples. Separation was achieved on a porous graphitic carbon (PGC) column without ion-pairing agent in the mobile phase. Low levels of the ion-pairing agent diethylamine (DEA) added to the reconstitution solution were necessary to prevent peak tailing of nucleoside triphosphates. The mass spectrometer, a triple quadrupole with an electrospray ionisation source, was operated in positive mode. Two multiple reaction monitoring (MRM) segments were programmed, each an internal standard. Extraction and separation of nucleoside mono- and triphosphates were obtained within 20 min. The total duration of a single run was 37 min. Calibration curves, performed with labelled nucleotides added to the sample matrix, ranged from 0.29 to 18.8 pmol injected for deoxyribonucleotides and from 3.9 to 3,156 pmol for ribonucleotides. Accuracy did not deviate more than −14.6 and 10.2 % from nominal values for all compounds at all levels. CV results were all lower than 17.0 % for the LLOQ level and 14.6 % for the other levels. Quality control (QC) samples were also in agreement with acceptance criteria, except for the lower QC of GMP. Ion suppression, matrix effect, extraction recoveries and stability were assessed. After validation, the method was applied to the evaluation of the effects of gemcitabine and hydroxyurea on nucleotide pools in Messa cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nucleotides are essential molecules in living organisms, playing roles as components of nucleic acids and co-substrates in enzymatic reactions, energy carriers, etc. The tight regulation of intracellular concentrations of nucleotides is important to maintain cellular homeostasis and involves a large number of enzymes. Indeed, modifications in these pools have been shown to be responsible for genetic instability and cell transformation [1]. In addition, intracellular nucleotides are in competition with active forms of nucleoside analogs used as anti-cancer, anti-viral and immunosuppressive therapy, thus modifying their activity. Therefore, the study of these endogenous compounds has become of great interest.
Determination of intracellular concentrations of nucleotides is challenging due to their low levels, the difference in concentrations between deoxyribonucleotides and ribonucleotides, their ionic nature and their chemical similarities. Nucleotides exhibited a poor retention on reverse phase high-performance liquid chromatography (HPLC) columns with conventional mobile phase. Therefore, many chromatographic methods based on anion exchange or ion-pairing (IP) mechanisms have been developed over the last years [2–5]. In IP chromatography, stationary phases are usually C18 or porous graphitic carbon (PGC). Anion exchange and IP methods allow good separation of nucleotides, but the use of high levels of salts or ion-pairing agents hamper with mass spectrometric detection [6]. To overcome this issue, methods using low levels of ion-pairing agent or no ion-pairing agent have been published. In this way, Zhao et al. added the ion-pairing agent only to the reconstitution solution to analyse the triphosphate metabolite of a nucleoside analog on a C18 column [6]. Thus, only low levels of ion-pairing agent were present in the ionisation source. Another solution is to replace the ion-pairing agent by an eluting ion as proposed by Jansen et al. who used ammonium bicarbonate to separate phosphate metabolites of gemcitabine on a PGC column [7]. Peifer et al. developed a liquid chromatography–tandem mass spectrometry (LC-MS/MS) method without using any ion-pairing agent to separate nucleosides and nucleotides [8]. However, they analysed only nucleoside monophosphates, while it is known that ion-pairing agent is particularly important for the separation of nucleosides triphosphates by decreasing peak tailing [9].
Although many methods relative to endogenous nucleotides separation and detection have been published [4, 9, 10], only a few described a complete analytical validation of the method [3, 11]. Most of the articles presented calibration, accuracy, precision, but no results on stability of nucleotides was reported [8, 12].
Cellular samples constitute complex analytical matrix with a large number of endogenous compounds. The first step of the analytical workflow for the study of intracellular nucleotides consists in a protein precipitation using various solvents such as 60 or 70 % methanol, trichloroacetic acid or perchloric acid (review in [13]). Thereafter, some authors perform a solid-phase extraction using anion exchange cartridge to isolate selectively nucleotides or phosphate metabolites of nucleoside analogs [3, 14–16]. However, this extraction step is time consuming and causes variability in repeatability and reproducibility. Online extraction enables automation and reduction of manual preparation steps. It also improves accuracy and precision [17]. Although the use of online extraction increases for several years, no publication applied to nucleotides has been published.
In this paper, we describe the development of an online coupling of anion exchange extraction to LC-MS/MS for the separation of nucleoside mono-, di- and triphosphates. This method, without ion-pairing agent in the mobile phase, allowed the quantification of ribonucleotides triphosphates (NTP), deoxyribonucleotides triphosphates (dNTP), ribonucleotides monophosphates (NMP) and deoxyribonucleotides monophosphates (dNMP). It was validated, and then applied on the evaluation of the effects of gemcitabine and hydroxyurea on nucleotide pools in Messa cells.
Material and methods
Chemicals and reagents
Standards (100 mM) of adenosine-13C10,15N5 5′-triphosphate (ATP13C,15N), 2′-deoxyadenosine-13C10,15N5 5′-triphosphate (dATP13C,15N), uridine-13C9,15N2 5′-triphosphate (UTP13C,15N), thymidine-13C10,15N2 5′-triphosphate (TTP13C,15N), cytidine-15N3 5′-triphosphate (CTP15N), 2′-deoxycytidine-13C9,15N3 5′-triphosphate (dCTP13C,15N), guanosine-13C10 5′-triphosphate (GTP13C), 2′-deoxyguanosine-13C10,15N5 5′-triphosphate (dGTP13C,15N) were purchased in solution from Sigma-Aldrich (St-Quentin-Fallavier, France). Standards (100 mM) of adenosine-13C10 5′-triphosphate (ATP13C) and cytidine-13C9 5′-triphosphate (CTP13C), used as internal standards, came also from Sigma-Aldrich. Solutions of 100-mM standards of adenosine-13C10,15N5 5′-monophosphate (AMP13C,15N) and thymidine-13C10,15N2 5′-monophosphate (TMP13C,15N) came from Eurisotop (Saint Aubin, France). 2′-deoxyadenosine-13C10,15N5 5′-monophosphate (dAMP13C,15N), uridine-15N2 5′-monophosphate (UMP15N), cytidine-15N3 5′-monophosphate (CMP15N), 2′-deoxycytidine-13C9,15N3 5′-monophosphate (dCMP13C,15N), guanosine-13C10,15N5 5′-monophosphate (GMP13C,15N), 2′-deoxyguanosine-13C10,15N5 5′-monophosphate (dGMP13C,15N) were purchased from Sigma-Aldrich. HPLC-grade acetonitrile, methanol, ammonia hydroxide aqueous solution (NH4OH), acetic acid, formic acid, diethylamine (DEA), hexylamine (HA) and hydroxyurea (HU) were obtained from Sigma-Aldrich. Gemcitabine (Gemzar®) came from Eli Lilly and Company (Neuilly-sur-Seine, France). Foetal bovine serum, streptomycin, penicillin, l-glutamine, phosphate buffered saline (PBS) and RPMI 1640 were obtained from Life technologies (Saint Aubin, France). Water filtered with a Millipore Milli-Q system (Molsheim, France) was used in all experiments.
Stock solutions
Stock solutions at 1 mM were prepared by dissolving individually the commercially labelled standards of NTP, dNTP, NMP and dNMP in ultrapure water and stored at −20 °C. Internal standard (IS) solutions of CTP13C and ATP13C at 20 μM were prepared in ultrapure water and stored at −20 °C.
Working solutions
Three levels of quality control (QC) samples and five levels of standard samples were freshly prepared by serial dilution of stock solutions in ultrapure water before each analysis/analytical run.
Preparation of cell extracts
Human uterine sarcoma cells (Messa) were grown in RPMI 1640 media containing l-glutamine, penicillin (200 UI/ml), streptomycin (200 μg/ml) and foetal bovine serum (10 %) at 37 °C in the presence of 5 % CO2. Cells at 80–90 % confluence were harvested by scraping in cold PBS, isolated by centrifugation (300 g, 5 min, 4 °C), washed with cold PBS and pelleted again by centrifugation. Cells were resuspended in cold methanol/water (v/v 70/30, 500 μl/2 million cells), vortexed for 20 s, snap-freezed in liquid nitrogen and stored at −80 °C until use. For applications, cells were exposed to hydroxyurea (2 mM) or gemcitabine (10 μM) the day after seeding and incubated for 24 h.
Sample processing
For each standard and QC sample, to 500 μl of cell lysate in methanol/water, 55 μl of working solution (or water if blank sample) and 10 μl of each internal standard solution were added. Samples were vigorously vortexed and then centrifuged for 10 min at 15,000g. Supernatant was evaporated to dryness under nitrogen at 37 °C. The residue was resuspended with 100 μl of 5-mM HA—0.5 % DEA in water and then centrifuged for 10 min at 15,000g. The supernatant was transferred to a 250-μl insert vial into an autosampler vial.
Instrumentation and experimental conditions
Online extraction and chromatography
Online solid-phase extraction was composed of two switching Rheodyne® valves and an extraction column (Oasis® WAX column (3.9 mm ×20 mm; 30 μm), (Waters, Guyancourt, France)). The chromatographic system consisted of a Surveyor AS® autosampler injector (Thermo Scientific, Courtaboeuf, France) and a Surveyor MS® quaternary pump (Thermo Scientific). The analytical column was a Hypercarb® column (2.1 mm × 100 mm; 5 μm, Thermo Scientific) and was thermostated at 30 °C. The autosampler tray was maintained at 5 °C and injection volume was 10 μl.
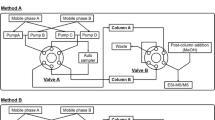
Two configurations are used during this analytical method (Fig. 1): a loading step in which mobile phase passed only through the extraction column, and an eluting step in which mobile phase transferred analytes from the extraction column to the analytical column. For the loading step, solvent was (B) water (200 μl/min, 2 min). For the eluting step, solvents were (A) NH4OH 0.25 % adjusted to pH10 with acetic acid, (B) and (C) acetonitrile (200 μl/min, 22 min). A stepwise gradient programme was used as shown in Fig. 2. Re-equilibration of Hypercarb® column was performed at a flow rate of 400 μl/min during 10 min, and re-equilibration of extraction column at a flow rate of 800 μl/min during 3 min. The entire run was achieved in 37 min. At the end of a series of analysis, extraction column was thoroughly washed with methanol (300 μl/min, 6 h) and analytical column with acetonitrile/water (80:20, v/v, 200 μl/min, 3 h).

Configuration of the online extraction system (EC extraction column, AC analytical column)

Online extraction and stepwise gradient programme. 1 valves in loading position, 2 valves in eluting position
Mass spectrometry
The mass spectrometer was a TSQ Quantum Ultra® triple quadrupole mass spectrometer (Thermo Scientific) equipped with an electrospray ionisation (ESI) source. It operated in positive ion multiple reaction monitoring (MRM) mode. Spray voltage and capillary voltage were set at 2,300 V and 350 °C, respectively. Argon was used as collision gas at 1.5 mTorr. The pressure of nitrogen sheath gas, auxiliary gas and sweep gas were maintained at 50, 10 and 2 units (arbitrary units). Scans of 30 ms were performed. A switching valve directed the eluate to waste during the first 9 min of the run and during re-equilibration step. Two segments were programmed. Cytidine monophosphate (CMP), cytidine triphosphate (CTP), deoxycytidine monophosphate (dCMP), deoxycytidine triphosphate (dCTP), uridine monophosphate (UMP) and uridine triphosphate (UTP) were detected in the first segment (from 9 to 13 min), and the other nucleotides in the second one (from 13 to 20 min).
The mass spectrometric fragmentation of each labelled NTP, dNTP, NMP and dNMP was studied for negative and positive ionisation. Solutions of each analyte at 0.1 mM in 5-mM HA—0.5 % DEA pH10 were infused at 10 μl/min and increased collision energies were applied. For these experiments, ion product mode was used.
Data were acquired using X-Calibur 2.0 software (Thermo Scientific).
Validation procedure
All validation procedure experiments were performed on Messa cells. Validation procedure has been realised according to recommendations of Shah et al. [18].
Calibration
To work in the same matrix as samples, quantification was performed by adding standard solutions of labelled nucleotides to cellular samples. Concentrations of endogenous nucleotides were calculated using calibration curves of the corresponding labelled nucleotides.
Calibration curves were obtained by spiking cell matrix with eight labelled nucleoside triphosphates and eight labelled nucleoside monophosphates at five different concentrations (Table 1). A calibration curve for each labelled analyte was obtained by quadratic regression analysis with 1/X weighting based on the peak area ratio of the analyte to its internal standard. A total of six calibrations curves were generated during the entire validation process. Calibration curves were built using LCquan® 2.5 (Thermo Fischer Scientific). Concentrations were back-calculated from the constructed calibration curve. Deviations from the nominal concentrations, expressed as accuracy, should be within ±20 % for the LLOQ and within ±15 % for other concentrations, with coefficient of variation values less than 20 and 15 %, respectively.
Accuracy and precision
For each labelled compound, between-run precision (BRP), within-run precision (WRP) and accuracy were calculated on three separated analytical runs by four replicates analysis of QC samples at three levels (Table 1). An estimate of BRP was obtained by one-way analysis of variance (ANOVA) for each test concentration using “run day” as a classification variable. The WRP was determined as \( \mathrm{WRP}=100\times \left(\sqrt{M{S}_{\mathrm{wit}}}/ GM\right) \). The BRP was estimated as \( \mathrm{BRP}=100\times \left(\sqrt{\left(M{S}_{\mathrm{bet}}-M{S}_{\mathrm{wit}}/n\right)}/ GM\right) \). MS wit, MS bet, n and GM represented the within-groups mean square, the between-groups mean square, the number of replicate observations within each run and the grand mean, respectively. These parameters were calculated using the software StatView for windows version 5.0 (SAS institute, Cary, USA). Accuracy was expressed as the percentage of deviation from the nominal concentration.
Selectivity
For all analytes, the lower limit of quantification (LLOQ) was chosen as the concentration of the lowest calibration sample. To evaluate selectivity, blank samples of Messa cells were checked for analyte and internal standard interfering peaks.
Matrix effect and ion suppression
The ion suppression was assessed by injecting a “blank” cellular sample during continuously post-column infusion of labelled nucleotides. Moreover, to investigate matrix effect, cellular and aqueous samples were spiked with the C5 level of labelled nucleotides and were prepared as described in “sample processing”. After online extraction and LC-MS/MS analysis, peak areas of each analyte from cellular samples were compared with those observed with aqueous samples which represented 100 % recovery.
Online extraction recovery
To investigate online extraction recovery, C1, C3 and C5 standard samples were prepared in triplicate for each level. Each of them was injected with and without online extraction device. Peak areas of analytes were compared and recoveries were calculated by using samples without online extraction as reference.
Stability
Pure solutions of labelled nucleotides were prepared as described above. The stability in the reconstitution solution was assessed after 12 h at 5 °C in the autosampler. Stability of cellular samples spiked with labelled nucleotides was studied by reinjection of standard samples after 6, 9 and 12 h maintained at 5 °C in the autosampler. Analytes were considered stable if the determined concentrations did not deviate more than ±15 % from the concentrations determined at time zero.
Carryover
Carryover was determined by injecting a blank sample after the highest calibration standard. It was expressed, in percentage, by (response of blank sample)/(response of calibration sample) × 100.
Results and discussion
Analytical development
Online extraction
The most common approaches for solid-phase extraction of nucleotides or phosphates metabolites of nucleoside analogs are based on anion exchange mechanism, using offline device [3, 14–16]. Online extraction assay development was based on our previous method using offline solid-phase extraction with Oasis® WAX cartridge [3]. Considering this report, we first assessed the effect of loading the sample onto online extraction column with 50-mM NH4OAc pH 4.5 and eluting the nucleotides with NH4OH 0.25 % pH 10 in order to use the anion exchange properties of the WAX column. Nucleoside monophosphates eluted first, then only a part of nucleoside triphosphates (about 20–30 %) eluted in very large peaks. Moreover, retention times were still increasing over injections, suggesting a distortion of WAX column properties that might be due to interactions between acetate and ionised piperazine groups of WAX column. To avoid interactions between salt and the stationary phase, formic acid was tested at 0.2 and 0.02 %. Nucleoside monophosphates eluted first, while only 10 % of nucleoside triphosphates eluted. Considering these observations, loading sample onto online extraction column in an aqueous mobile phase was tested. Under these conditions, only a small amount of NH4OH 0.25 % pH 10 was necessary to elute all the nucleotides. A preconditioning extraction column step with formic acid was also tested, but did not bring any improvement. Therefore, the procedure with the loading step under water was selected for further development.
Online extraction column played a role on the chromatographic separation of nucleotides. First, extraction column separated nucleoside monophosphates from nucleoside triphosphates. Nucleoside monophosphates eluted the extraction column at about 4 min while nucleoside triphosphates eluted in large peaks ranging from 6 to 9 min. However, it seems to have no effect on the order in which nucleotides eluted the PGC column. In fact, the order was similar with or without online extraction column. Moreover, we observed that online extraction column improved peak shape.
To our knowledge, it is the first report of an analytical method for quantification of nucleoside mono- and triphosphates using online extraction. Kuklenyik et al. reported an online coupling of anion exchange extraction and ion-pair chromatography for the measurement of triphosphate metabolites of anti-viral reverse transcriptase inhibitors using a different device [19]. Online coupling of extraction and chromatography presents the advantages to be automatable and less time consuming than offline solid-phase extraction. However, some issues arise from the combination with LC-MS/MS. The main difficulty is the compatibility of solvents between the extraction step, the chromatographic separation and the MS detection.
Optimization of liquid chromatography
Ion-pairing agent
Several methods for the quantification of nucleotides used an ion-pairing agent in the mobile phase [3–5]. However, ion-pairing agents are known to reduce sensitivity and to be a cause of ion suppression [6, 20]. Similar to what has been reported in previous studies [6, 7], our first requirement was to reduce or even eliminate the exposure of the ion-pairing agent in the LC-MS/MS system.
One solution is to add the ion-pairing agent only to the reconstitution solution. It has been successfully reported by Liu et al. in the detection of a tetra-phosphate and by Zhao et al. in the quantification of a triphosphate metabolite of a nucleoside reverse transcriptase inhibitor [6, 21]. Jansen et al. proposed a method for the separation of nucleoside analogs and their mono-, di- and triphosphates on a PGC column without an ion-pairing agent [7]. They reported the interest of introducing an eluting ion such as bicarbonate in the mobile phase. However, they encountered loss of separation capabilities of the column probably due to a strong affinity of bicarbonate with the PGC column. Considering difficulties presented by Jansen et al., we first evaluated the effect of adding 5-mM HA—0.5 % DEA to the reconstitution solution. Nucleoside mono- and triphosphates were well separated and peak shapes were satisfactory. The latter were altered in the absence of one of the two compounds proving the importance of HA and DEA. Improvement of peak shapes was more evident for DEA since peak tailing appeared for nucleoside triphosphates in the absence of DEA (Fig. 3). It was also observed that ion-pairing agent did not affect the retention times (data not shown). The reduction of peak tailing by DEA was previously observed for nucleoside di- and triphosphates [9]. According to the literature, peak tailing results from interactions between phosphate compounds and stainless steel [22–24]. Therefore, triphosphate compounds are more affected than diphosphates and monophosphates [9]. In our method, only a small amount of DEA was necessary to prevent peak tailing of nucleoside triphosphates.

Effect of DEA in the reconstitution solution on tailing factor. Tailing factor determined according to the US Pharmacopeia
Mobile phase
Due to the online extraction coupling, NH4OH 0.25 % pH 10 was needed in the mobile phase. Ten percent were sufficient to elute the nucleotides off the extraction column. An increase in NH4OH (from 10 to 30 %) had no effect on retention times, but resulted in a decrease in MS responses. Thus, the proportion of NH4OH 0.25 % pH 10 was set to 10 % of the mobile phase. A binary gradient including water and acetonitrile completed the mobile phase. Under these conditions, nucleotides separation depended on the nature of the base with elution in the following order: cytosine, uridine, thymidine, guanosine and adenosine. Concerning the level of phosphorylation of a given nucleoside, monophosphates, diphosphates and triphosphates forms were eluted successively. Finally, as for the difference on the sugar moiety, deoxyribonucleotides eluted after ribonucleotides for the same base and level of phosphorylation (Fig. 4). Conditions of mobile phase were optimised to expand the range of retention times in order to avoid competition on the ionisation step between the different nucleotides present in the electrospray source. It was observed that maintaining acetonitrile at 7 % of the mobile phase at the beginning of the gradient programme increased retention times and improved separation of analytes. Thus, the gradient programme began with the following conditions during 4 min: 10 % NH4OH 0.25 % pH 10, 83 % water and 7 % acetonitrile. Then, a gradient with a slope of 2.5 %/min provided a sufficient separation (Fig. 2). Finally, under the optimised conditions, the first nucleotide (CMP) eluted at 10 min and the last (deoxyadenosine triphosphate (dATP)) at 16.5 min, with good peak shapes (Fig. 5). These conditions allowed two segments with time-programmed MRM of analytes. A period of 4 min with 80 % acetonitrile was introduced after elution of nucleotides to clean matrix elements on the two columns. In order to reduce re-equilibration time, the flow rate was increased to 400 μl/min for the PGC column and 800 μl/min for the extraction column. No modification of the retention times for the next analysis was observed. Moreover, this procedure brought a significant advantage compared to other methods using a PGC column without ion-pairing agent in the mobile phase which required re-equilibration time between 30 min and 2 h [7, 25, 26].

MRM chromatograms of AMP, ADP, ATP, dAMP, dADP and dATP in a cellular sample

MRM chromatogram of labelled nucleotides of a spiked cellular sample
The effect of methanol on the retention of nucleotides was also evaluated by replacing acetonitrile by methanol. Pyrimidine nucleotides were eluted later, whereas purine nucleotides were trapped on the PGC column. A long cleaning step (2 h) was necessary to elute them off the PGC column. Isopropanol was proposed by Zhao et al. as a mobile phase modifier. In their study, 10 % isopropanol in acetonitrile improved peak shape and increased peak height of a triphosphate metabolite of a nucleoside reverse transcriptase inhibitor [6]. Under our conditions, replacing acetonitrile by a mixture of isopropanol/acetonitrile (10:90, v/v) resulted in an increase in peak widths, and it was therefore not included in our mobile phase.
The ruggedness of methods for the separation of nucleotides caused problems for many authors. After one run, Jansen et al. observed a deterioration of separation capabilities of the PGC [7]. To restore them, an acidic solution was injected after each run. Moreover, they reported a requirement of 10–20 gradient runs to reach stable retention times within an analytical run. Using a C18 column and DEA in the reconstitution solution, Zhao et al. also described difficulties concerning assay ruggedness [6]. They were faced with loss of sensitivity, peak shape deterioration and increased carryover. We tested the ruggedness by injecting 20 consecutive cellular samples. Neither change in retention time nor deterioration of peak shapes was observed, and only two injections were necessary to reach stable retention times. Therefore, this method is applicable to a sequence of at least 20 samples. Based on our practice, we decided to clean the PGC column each day with acetonitrile/water (80:20, v/v; 200 μl/min) during 3 h. We also cleaned the extraction column by methanol during a longer time (6 h) every day. This long-time cleaning was necessary to avoid a deterioration of peak shapes of nucleoside triphosphates. If a large number of samples have to be analysed consecutively, the disadvantage of long cleaning can be easily bypassed by using alternatively two sets of columns.
Finally, extraction and separation of nucleoside mono-, di- and triphosphates were obtained within 20 min. With the re-equilibration steps, the total duration of a run is 37 min, representing a significant time saving compared to methods having chromatographic run times higher than 60 min [3, 4, 10, 27].
Internal standards
Nucleotides differently labelled from those used as standards were chosen. CTP13C was used as internal standard for nucleotides eluted during the first segment, and ATP13C for nucleotides eluted during the second segment.
Mass spectrometry
Positive and negative ion modes were tested in order to select the most relevant conditions for each compound. Experiments were conducted on the labelled nucleotides used as standards and mass spectrometric fragmentations were confirmed with unlabelled nucleotides.
As previously described in negative ion mode, mass fragmentation of NMP and dNMP (except deoxyadenosine monophosphate (dAMP)) showed a common pattern at m/z 79 corresponding to the monophosphate moiety [PO3]− [10, 12]. Thus, adenosine monophosphate (AMP) and deoxyguanosine monophosphate (dGMP) present the same transition (m/z 346 > 79), requiring a chromatographic separation between the two nucleotides. dAMP13C15N fragmented in a main product ion at m/z 144 corresponding to the base. Petroselli et al. reported a similar MS/MS spectrum for dAMP showing a major product ion corresponding to adenine and the monophosphate moiety as a secondary product ion [28]. In the literature, most studies relative to the analysis of nucleoside monophosphates by mass spectrometry used the negative mode to monitor AMP, CMP, UMP and guanosine monophosphate (GMP) [10, 12, 27]. In positive mode, we observed that the mass fragmentation of AMP, dAMP, CMP, dCMP, GMP and dGMP revealed a single product ion at m/z corresponding to their base. These data confirmed those reported by Peifer et al. and by Yang et al. for AMP and GMP [8, 29]. For UMP and thymidine monophosphate (TMP) two main fragments at m/z 97 and m/z 81 were observed, respectively. The fragmentation pathway was studied using different labelled compounds. UMP15N exhibited the same fragment at m/z 97 than the unlabelled UMP. UMP13C,15N and TMP13C,15N, with a 13C labelling occurring on five carbons of the sugar moiety showed a fragment at m/z 102 and 86, respectively, representing a shift of 5 amu with the unlabelled UMP and TMP. Thus, the two fragments were attributed to the sugar moiety. One study using an ion trap system proposed a different MS/MS spectrum of UMP showing a main product ion at m/z 213 corresponding to [M-uridine]+ [29]. Since DEA was present in the solution containing nucleotides, [M + DEA]+ adducts were observed for all NMP and dNMP. Responses of [M + DEA]+ adducts represented less than 7 % as compared to responses of [M + H]+ (Table 2). The fragmentation of [M + DEA]+ appeared at low-collision energy (5 V) and produced a product ion at m/z corresponding to [M + H]+.
Considering the presence of negative charges on phosphate groups, the use of negative mode appeared to be particularly relevant for nucleoside triphosphates. As reported in the literature, in negative mode, nucleoside triphosphates fragmented in a most abundant product ion at m/z 159, corresponding to the pyrophosphate fragment [HP2O6]− [3, 10, 30]. In positive mode, nucleoside triphosphates formed a unique fragment corresponding to their base, except for UTP and thymidine triphosphate (TTP). Similar to UMP and TMP, UTP and TTP showed a more abundant product ion corresponding to the sugar moiety. [M + DEA]+ adducts were also observed, with a fragmentation appearing at low collision energy. Their responses do not exceed 7 % in comparison to responses of [M + H]+ (Table 2).
Thereby, in negative mode, fragmentation leads to common patterns at m/z 79 and m/z 159 for nucleoside mono- and triphosphates, respectively. Thus, for nucleotides differing from 1 Da (TMP, CMP, UMP and TTP, CTP, UTP), a chromatographic separation is necessary to avoid bias due to isotope distribution. As mentioned above, this inconvenience does not exist in positive mode since nucleotides present specific patterns.
For nucleoside mono- and triphosphates, (except TTP), responses were clearly higher in positive mode than in negative mode (Table 2). As reported previously by Fung et al. and by Pruvost et al., a better ionisation of TTP in negative mode than in positive mode was observed [31, 32]. Fung et al. proposed the role played by the basicity of the nucleobase to explain a less efficient ionisation of TTP in positive mode [31]. The hypothesis that nucleotides with a weaker base had more efficient ionisation in the negative mode was recently confirmed by Quinn et al. [33]. Pruvost et al. emphasised that nucleoside triphosphates with amino-substituted base were better ionised than nucleoside triphosphates with only nitrogens in the heterocyclic base [32].
In the absence of DEA, nucleoside mono- and triphosphates were also ionised in positive mode. The effects of DEA on these responses differed between nucleoside mono- and triphosphates. When adding DEA to the reconstitution solution, ionisation of nucleoside monophosphates appeared 15–20 % less efficient as compared to the absence of DEA. For nucleoside triphosphates, only a slight increase (10 %) in responses was observed. Considering the great interest of DEA on nucleotides separation, DEA was still added to the reconstitution solution.
Cordell et al. reported high background interference from the protonated DMHA and a lack of sensitivity due to an increased distribution of the total ion content in positive mode [12]. Under our conditions, signal to noise ratios were higher in positive mode than in negative mode for all analytes (except TTP). No high background interference was seen in the presence of DEA. This could be explained by the presence of ion pairing agent only in the injected solution, and not in the mobile phase.
Finally, in the present study, considering intensities, selectivity of main product ions and signal to noise ratios, positive mode was selected (Fig. 6). Although TTP exhibited a better response in negative mode, positive mode was also selected for this analyte because in this configuration, a shift between the two modes required time and leaded to a decrease of the analysis time spent for detection of each compound. Once ionisation mode and transitions have been chosen, collision energy was optimised for each analyte (Table 3).

MS fragmentation pathways of endogenous nucleotides in positive mode. R = H4P3O10 for nucleoside triphosphates; R = H2PO4 for nucleoside monophosphates. Deoxyribonucleotides forms are represented without the OH group in dashed box
Method validation
Validation has been performed for nucleoside mono- and triphosphates using commercially available labelled nucleotides. Although, nucleotide diphosphates were clearly detected with the present assay, to our knowledge, no labelled nucleoside diphosphate is commercially available. Therefore, their quantification may be performed using calibration curves of nucleoside mono- or triphosphates according to the range of concentrations expected for nucleoside diphosphates.
Calibration
A total of six calibration curves were generated during the entire validation process. Accuracy did not deviate more than −14.6 and 10.2 % from nominal values for all compounds at all levels. CV results were all lower than 17.0 % for the LLOQ level and 14.6 % for the other levels. Therefore, all results were accepted (Table 4).
Accuracy and precision
Table 5 summarises WRP, BRP and accuracy of QC samples at three levels. Values were within ±20 % at the lower QC level and ±15 % at the other levels, and thus, in agreement with acceptance criteria for all nucleotides expect for GMP. WRP and accuracy of GMP were higher than 20 % at the lower QC level. These results could be explained by the concave shape of the calibration curve of GMP with a weak slope value at low levels leading to a poor sensitivity.
Selectivity and specificity
For TMP, dCMP, dGMP, dATP, TTP, dCTP and deoxyguanosine triphosphate (dGTP), different range of concentrations were tested and the LLOQ was set at the concentration corresponding to the lowest standard. For the other nucleotides, the LLOQ were in the same range of quantity injected (0.3 to 1 pmol). However, for these compounds, the ranges of standard curves were adjusted according to their concentrations into cells.
No interfering peaks were observed in the chromatogram from a blank sample of Messa cells at the retention times of each analyte and internal standard. An example of chromatogram is presented in Fig. 7 for the two nucleotides having the lowest standard values (dCMP and TMP).

MRM chromatograms of dCMP13C,15N and TMP13C,15N in a blank sample and b spiked cellular sample
Matrix effect and ion suppression
No ion suppression due to cellular constituents was observed by post-column infusion of labelled nucleotides and internal standards. The evaluation of the matrix effect was carried out by comparison of responses of cellular and aqueous samples. The matrix effect was lower than 10 % for all analytes, except for CMP15N, dCMP13C,15N and GMP13C,15N where it was measured at 25, 29 and 31 %, respectively.
Online extraction recovery
Online extraction recoveries were independent of nucleotide concentrations, and ranging from 95 to 105 % for AMP, dAMP, GMP, dGMP, ATP, dATP, UTP, TTP, CTP, dCTP, guanosine triphosphate (GTP) and dGTP. For UMP, values were between 85 and 95 %, and for TMP, CMP and dCMP between 70 and 85 %. All values were much higher than the solid-phase extraction recoveries of NTP and dNTP reported earlier [3]. After a simple protein precipitation, recoveries ranged from 60 to 100 % [12, 13, 34].
Stability
Stability of nucleoside mono- and triphosphates extracted from pure solutions and maintained during 12 h at 5 °C in the reconstitution solution was acceptable since responses did not deviate more than ±15 % from initial values.
For nucleotides extracted from Messa cells, stability at 5 °C in the reconstitution solution depended on the nucleotide. Responses of ATP, dATP, UTP, TTP, CTP, dCTP, GTP, AMP, dAMP, UMP, TMP, CMP and dCMP were stable during the entire period of the test (12 h). The majority of nucleotides derived from guanosine presented instability of the responses in our conditions of analysis. dGTP showed increasing responses over time: +40 % at 6 h and +60 % at 9 and 12 h. Responses of GMP decreased dramatically: −40 % at 6 h and −70 % at 9 h. The decrease was significant from 2 h (−16 %). For dGMP, the decrease was slower: −20 % at 6 h, −30 % at 9 h and −70 % at 12 h. The first hypothesis advanced to explain these results was phosphorylation or dephosphorylation reactions occurring during storage on the autosampler. This hypothesis was not retained after studying the stability of responses of nucleoside diphosphates during storage on the autosampler. Only a few data are available on the stability of endogenous nucleotides. In similar conditions, Cohen et al. described a stability of nucleoside triphosphates, dGTP included, during 40 h at 5 °C [3]. Their method coupled an offline extraction with LC-MS/MS analysis, so stability was tested on post-extracted samples. We hypothesised that instability of responses under our conditions was caused by an endogenous compound still present in the autosampler vial because extraction was not yet performed. In practice, procedure must be adapted to the nucleotide analysed. For example, if GMP is quantified, the dry residue should be resuspended extemporaneously before injection.
Carryover
Carryover was less than 2 % of the area for ATP and GTP, and less than 1 % of the area for the other nucleotides. Therefore, carryover of the analytes and internal standards was acceptable.
Applications
Messa cells were exposed to gemcitabine (10 μM) or hydroxyurea (HU) (2 mM) for 24 h, the day after seeding. Endogenous nucleotides were quantified and compared to untreated Messa cells and to data published in the literature. After treatment with gemcitabine, all nucleoside monophosphates and triphosphates levels increased significantly, except dATP which was unaffected (Fig. 8). Such results are in agreement with data reported by Van Moorsel et al. for nucleoside triphosphates [35]. Treatment of Messa cells with HU resulted in a significant decrease of dATP, as reported by several teams [36, 37]. We also observed a significant increase of dCTP and stable levels of TTP and dGTP (Fig. 9); for these three dNTP, results published after treatment by HU are discordant [36–39]. It is known that the effects of HU on dNTP are complex and depend on the cell lines [36, 37]. Therefore, the data obtained from our experiments are consistent with the results expected based on the literature, proving the suitability of the present assay for the study of nucleotide pools.

Effects of gemcitabine on nucleotide pools in Messa cells. Cells were exposed to gemcitabine 10 μM for 24 h. Values are means ± SD of at least five samples. *Significantly different from untreated cells (p < 0.05)

Effects of hydroxyurea on nucleotide pools in Messa cells. Cells were exposed to hydroxyurea 2 mM for 24 h. Values are means ± SD of at least five samples. +<0.1 pmol injected; *Significantly different from untreated cells (p < 0.05)
Conclusion
In this study, a LC-MS/MS method without ion-pairing agent in the mobile phase was developed for the analysis of nucleotides. For the first time, the analytical method was coupled with an online extraction on a weak-anion exchange column. This method allowed the separation and the detection of nucleoside mono-, di- and triphosphates, deoxyribonucleotides and ribonucleotides. It also permitted the quantification of nucleoside mono- and triphosphates. As no ion-pairing agent was present in the mobile phase, the method was more rugged than other methods. Other advantage was provided by the online extraction which was less time consuming. The method was fully validated, except for GMP, so it can be applied for the investigation of nucleotide pools in cellular samples.
References
Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145:435–446
Jansen RS, Rosing H, de Wolf CJ, Beijnen JH (2007) Development and validation of an assay for the quantitative determination of cladribine nucleotides in MDCKII cells and culture medium using weak anion-exchange liquid chromatography coupled with tandem mass spectrometry. Rapid Commun Mass Spectrom 21:4049–4059
Cohen S, Jordheim LP, Megherbi M, Dumontet C, Guitton J (2010) Liquid chromatography methods for the determination of endogenous nucleotides and nucleotide analogs used in cancer therapy: a review. J Chrom B 878:1912–1928
Zhang W, Tan S, Paintsil E, Dutschman GE, Gullen EA, Chu E, Cheng YC (2011) Analysis of deoxyribonucleotide pools in human cancer cell lines using a liquid chromatography coupled with tandem mass spectrometry technique. Biochem Pharmacol 82:411–417
Yamaoka N, Kudo Y, Inazawa K, Inagawa S, Yasuda M, Mawatari KI, Nakagomi K, Kaneko K (2010) Simultaneous determination of nucleosides and nucleotides in dietary foods and beverages using ion-pairing liquid chromatography–electrospray ionization-mass spectrometry. J Chrom B 878:2054–2060
Zhao Y, Liu G, Liu Y, Yuan L, Hawthorne D, Shen JX, Guha M, Aubry A (2013) Improved ruggedness of an ion-pairing liquid chromatography/tandem mass spectrometry assay for the quantitative analysis of the triphosphate metabolite of a nucleoside reverse transcriptase inhibitor in peripheral blood mononuclear cells. Rapid Commun Mass Spectrom 27:481–488
Jansen RS, Rosing H, Schellens JHM, Beijnen JH (2009) Retention studies of 2′-2′-difluorodeoxycytidine and 2′-2′-difluorodeoxyuridine nucleosides and nucleotides on porous graphitic carbon: development of a liquid chromatography–tandem mass spectrometry method. J Chromatogr A 1216:3168–3174
Peifer S, Schneider K, Nürenberg G, Volmer DA, Heinzle E (2012) Quantitation of intracellular purine intermediates in different Corynebacteria using electrospray LC-MS/MS. Anal Bioanal Chem 404:2295–2305
Xing J, Apedo A, Tymiak A, Zhao N (2004) Liquid chromatographic analysis of nucleosides and their mono-, di- and triphosphates using porous graphitic carbon stationary phase coupled with electrospray mass spectrometry. Rapid Commun Mass Spectrom 18:1599–1606
Tuytten R, Lemière F, Van Dongen W, Esmans EL, Slegers H (2002) Short capillary ion-pair high performance liquid chromatography coupled with electrospray (tandem) mass spectrometry for the simultaneous analysis of nucleoside mono-, di- and triphosphates. Rapid Commun Mass Spectrom 16:1205–1215
Wang J, Lin T, Lai J, Cai Z, Yang MS (2009) Analysis of adenosine phosphates in HepG-2 cell by a HPLC–ESI-MS system with porous graphitic carbon as stationary phase. J Chrom B 877:2019–2024
Cordell RL, Hill SJ, Ortori CA, Barrett DA (2008) Quantitative profiling of nucleotides and related phosphate-containing metabolites in cultured mammalian cells by liquid chromatography tandem electrospray mass spectrometry. J Chrom B 871:115–124
Cohen S, Megherbi M, Jordheim LP, Lefebvre I, Perigaud C, Dumontet C, Guitton J (2009) Simultaneous analysis of eight nucleoside triphosphates in cell lines by liquid chromatography coupled with tandem mass spectrometry. J Chrom B 877:3831–3840
Darque A, Valette G, Rousseau F, Wang LH, Sommadossi JP, Zhou XJ (1999) Quantitation of intracellular triphosphate of emtricitabine in peripheral blood mononuclear cells from human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 43(9):2245–2250
Font E, Rosario O, Santana J, García H, Sommadossi JP, Rodriguez JF (1999) Determination of zidovudine triphosphate intracellular concentrations in peripheral blood mononuclear cells from human immunodeficiency virus-infected individuals by tandem mass spectrometry. Antimicrob Agents Chemother 43(12):2964–2968
Robbins BL, Waibel BH, Fridland A (1996) Quantitation of intracellular zidovudine phosphates by use of combined cartridge-radioimmunoassay methodology. Antimicrob Agents Chemother 40(11):2651–2654
Chen L, Wang H, Zeng Q, Xu Y, Sun L, Xu H, Ding L (2009) Online coupling of solid-phase extraction to liquid chromatography—a review. J Chromatogr Sci 47:614–623
Shah VP, Midha KK, Findlay JWA, Hill HM, Hulse JD, McGilveray IJ, McKay G, Miller KJ, Patnaik RN, Powell ML, Tonelli A, Viswanathan CT, Yacobi A (2000) Bioanalytical method validation—a revisit with a decade of progress. Pharm Res 17(12):1551–1557
Kuklenyik Z, Martin A, Pau CP, Holder A, Youngpairoj AS, Zheng Q, Cong ME, Garcia-Lerma JG, Heneine W, Pirkle JL, Barr JR (2009) On-line coupling of anion exchange and ion-pair chromatography for measurement of intracellular triphosphate metabolites of reverse transcriptase inhibitors. J Chrom B 877:3659–3666
Vela JE, Olson LY, Huang A, Fridland A, Ray AS (2007) Simultaneous quantitation of the nucleotide analog adefovir, its phosphorylated anabolites and 2-deoxyadenosine triphosphate by ion-pairing LC/MS/MS. J Chrom B 848:335–343
Liu DY, Cojocaru L (2010) The determination of a tetra-phosphate compound in rat plasma. LC/MS/MS AAPS Annual Meeting and Exposition, New Orleans, LA, November 2010
Tuytten R, Lemière F, Witters E, Van Dongena W, Slegers H, Newton RP, Van Onckelen H, Esmans EL (2006) Stainless steel electrospray probe: a dead end for phosphorylated organic compounds? J Chromatogr A 1104:209–221
Jansen RS, Rosing H, Schellens JHM, Beijnen JH (2011) Mass spectrometry in the quantitative analysis of therapeutic intracellular nucleotide analogs. Mass Spec Rev 30:321–343
Wakamatsu A, Morimoto K, Shimizu M, Kudoh S (2005) A severe peak tailing of phosphate compounds caused by interaction with stainless steel used for liquid chromatography and electrospray mass spectrometry. J Sep Sci 28(14):1823–1830
Antonopoulos A, Favetta P, Helbert W, Lafosse J (2007) On-line liquid chromatography–electrospray ionisation mass spectrometry for κ-carrageenan oligosaccharides with a porous graphitic carbon column. J Chromatogr A 1147:37–41
Vainchtein LD, Rosing H, Schellens JHM, Beijnen JH (2010) A new validated HMPL-MS/MS method for the simultaneous determination of the anti-cancer agent capecitabine and its metabolites: 5′-deoxy-5-fluorouridine, 5-fluorouracil and 5′-fluorodihydrouracil, in human plasma. Biomed Chromatogr 24:374–386
Yamaoka N, Inazawa K, Inagawa S, Yasuda M, Mawatari K, Nakagomi K, Fujimori S, Yamada Y, Kaneko K (2011) Simultaneous determination of purine and pyrimidine metabolites in hprt-deficient cell lines. Nucleosides Nucleotides Nucleic Acids 30:1256–1259
Petroselli G, Erra-Balsells R, Cabrerizo FM, Lorente C, Capparelli AL, Braun AM, Oliveros E, Thomas AH (2007) Photosensitization of 2′-deoxyadenosine-5′-monophosphate by pterin. Org Biomol Chem 5:2792–2799
Yang FQ, Li DQ, Feng K, Hu DJ, Li SP (2010) Determination of nucleotides, nucleosides and their transformation products in Cordyceps by ion-pairing reversed-phase liquid chromatography–mass spectrometry. J Chromatogr A 1217:5501–5510
Coulier L, Gerritsen H, van Kampen JJA, Reedijk ML, Luider TM, Osterhaus ADME, Gruters RA, Brüll L (2011) Comprehensive analysis of the intracellular metabolism of antiretroviral nucleosides and nucleotides using liquid chromatography–tandem mass spectrometry and method improvement by using ultra performance liquid chromatography. J Chrom B 879:2772–2782
Fung EN, Cai Z, Burnette TC, Sinhababu AK (2001) Simultaneous determination of Ziagen and its phosphorylated metabolites by ion-pairing high-performance liquid chromatography–tandem mass spectrometry. J Chrom B 754:285–295
Pruvost A, Théodoro F, Agrofoglio L, Negredo E, Bénech H (2008) Specificity enhancement with LC-positive ESI-MS/MS for the measurement of nucleotides: application to the quantitative determination of carbovir triphosphate, lamivudine triphosphate and tenofovir diphosphate in human peripheral blood mononuclear cells. J Mass Spectrom 43:224–233
Quinn R, Basanta-Sanchez M, Rose RE, Fabris D (2013) Direct infusion analysis of nucleotide mixtures of very similar or identical elemental composition. J Mass Spectrom 48:703–712
Shi G, Wu JT, Li Y, Geleziunas R, Gallagher K, Emm T, Olah T, Unger S (2002) Novel direct detection method for quantitative determination of intracellular nucleoside triphosphates using weak anion exchange liquid chromatography tandem mass spectrometry. Rapid Commun Mass Spectrom 16:1092–1099
Van Moorsel CJA, Bergman AM, Veerman G, Voorn DA, Ruiz van Haperen VWT, Kroep JR, Pinedo HM, Peters GJ (2000) Differential effects of gemcitabine on ribonucleotide pools of twenty-one solid tumor and leukaemia cell lines. Biochim Biophys Acta 1474:5–12
Golkar SO, Czene S, Gokarakonda A, Haghdoost S (2013) Intracellular deoxyribonucleotide pool imbalance and DNA damage in cells treated with hydroxyurea, an inhibitor of ribonucleotide reductase. Mutagenesis 28:653–660
Hakansson P, Hofer A, Thelander L (2006) Regulation of mammalian ribonucleotides reduction and dNTP pools after DNA damage and in resting cells. J Biol Chem 281:7834–7841
Bianchi V, Pontis E, Reichard P (1986) Changes of deoxyribonucleoside triphosphate pools induced by hydroxyurea and their relation to DNA synthesis. J Biol Chem 261(34):16037–16042
Collins A, Oates D (1987) Hydroxyurea: effects on deoxyribonucleotide pool size correlated with effects on DNA repair on mammalian cells. Eur J Biochem 169:299–305
Acknowledgments
We would like to thank E. Cros for providing the Messa cells, and B. Vignal and S. Coste for their technical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Machon, C., Jordheim, L.P., Puy, JY. et al. Fully validated assay for the quantification of endogenous nucleoside mono- and triphosphates using online extraction coupled with liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 406, 2925–2941 (2014). https://doi.org/10.1007/s00216-014-7711-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-7711-1