
Abstract
Three hydroxypropyl methylcellulose samples (HPMC1–3, DSMe = 1.45, 1.29, and 1.36; MSHP = 0.28, 0.46, and 0.84) were analyzed with respect to their methyl and hydroxypropyl substitution pattern in the polymer chains. Ionization yield of HPMC oligomers in electrospray ionization ion trap mass spectrometry (ESI-IT-MS) is strongly influenced by the hydroxypropyl pattern. Therefore, a sample derivatization procedure, as well as suitable measurement conditions that enable relative quantification were elaborated. Analysis was performed by negative ESI-IT-MS after per(deutero)methylation, partial depolymerization, and reductive amination with m-aminobenzoic acid. Measurement parameters like solvent, trap drive, and voltages of the ion transportation unit were studied with regard to the suitability for quantitative evaluation. Using direct infusion of the samples, strong influence of trap drive and octopole settings was observed. Optimized measurement conditions were used for the determination of the HP pattern of the permethylated samples by direct infusion. The methyl pattern was determined from the perdeuteromethylated samples by high-performance liquid chromatography–electrospray tandem mass spectrometry. For HPMC1, substituents were both found to fit the random distribution model. The other two samples showed pronounced heterogeneity which could be interpreted in more detail by extracting methyl subpatterns depending on the number of HP groups.

Determination of the substituent distribution in hydroxypropyl methylcellulose oligomers by ESI-IT-MS (negative mode) after labeling with m-aminobenzoic acid
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hydroxypropyl methylcelluloses (HPMC, 1) are mixed cellulose ethers with a wide field of application in food, cosmetics, building materials, and industrial use [1]. In pharmaceutical products, HPMC is employed for controlled drug release [2]. Industrial production of HPMC is performed in a slurry process in which alkoxylation with methyl (Me) oxirane is carried out prior to or simultaneously with methylation with MeCl. Beside the hydroxyl functions of the cellulose, OH of the hydroxypropyl (HP) substituent can also react, and is partly methylated, while tandem reaction with oxirane are quite unlikely for HPMCs with a low molar degree of hydroxypropyl substitution (MSHP) of 0.1–0.4. The typical range for commercially available HPMCs is 0.1 to 1.0 for MSHP and 1.3 and 2.1 for degree of Me substitution (DSMe) [1].

Depending on the reaction conditions, the products differ in physicochemical properties like solubility or gelling and flocculation point [3, 4]. However, the characterization of these mixed ethers on molecular level is challenging because of the large variety of substitution patterns already in the glucosyl unit. Beside the average DS and the regioselectivity of substitution, also the substituent pattern along and over the polymer chains is important for the product properties [5, 6].
If uniform activation of the cellulose (the starting material) has been achieved, substitution reaction proceeds in a random manner with respect to the macromolecular level as it has been observed for Me cellulose [7, 8]. Heterogeneities among the polymer chains (first order) and within the chains (second order heterogeneity) can be detected by oligomer analysis and comparison of the experimentally determined substitution pattern with the distribution calculated for a random model [9, 10].
For the determination of the monomer composition of cellulose ethers, several chromatographic or electrophoretic methods have been applied [11–14]. In order to determine the substituent pattern along the polymer backbone, mass spectrometry (MS) is the method of choice because it allows simultaneous detection of a high number of various analytes in a wide mass range. Prior to mass spectrometric analysis, the polymer has to be partially depolymerized. To obtain representative results for all oligomers, the cleavage must proceed randomly without preferred cleavage of glycosidic linkages of a certain substitution pattern. For partial depolymerization of MC, free OH groups are isotopically labeled with Me-d 3 to obtain chemical uniformity and still maintain the structural information about the substitution pattern. However, because of the complexity of substituents in HPMC, such chemical uniformity cannot be achieved. Nevertheless, free OH groups are fully protected prior to analysis for observing the HP pattern independent of the degree of methylation, and at the same time enhancing signal intensity for the determination of the HP pattern (permethylation). However, to enable the analysis of the Me pattern, perdeuteromethylation is applied.
For mass spectrometric analysis of polysaccharides, soft ionization methods like electrospray ionization (ESI) and matrix- assisted laser desorption/ionization (MALDI) are widely applied [15]. However, the cation complexation ability of oligosaccharides strongly depends on the chemistry and number of substituents, and the resulting ion intensity and relative intensities do not reflect the real composition. Modification of free OH groups with MeI (or MeI-d 3) diminishes chemical differences, and additional introduction of a permanently charged tag improves comparability of molar ion yields [16].
The substituent distribution in HPMC has been analyzed successfully in the past by means of MALDI-ToF-MS after labeling of the analytes with propylamine and subsequent quaternization [17], but the labeling procedure and work up steps were time consuming and prone to sample loss and side reactions, and ESI-MS did not give quantifiable results. Recently, other labeling procedures have been elaborated [18, 19] and applied to MC by our group [8]. However, quantitative electrospray ionization ion trap mass spectrometry (ESI-IT-MS) of mixed cellulose ethers requires a more detailed insight in the effect of different measurement parameters. Sample treatment and sample concentration, as well as various parameters of the mass analyzer (voltages of ion transportation and ion trap (IT) influence the quality of the recorded spectra and need to be optimized [20–23]. In the application of (HPLC-)ESI-MS to single analytes in a complex matrix (e.g., in pesticide analytics), measurement parameters for the particular ion can be optimized [24, 25]. The other extreme is the determination of the molar mass distribution of (synthetic) polymers. MS plays an increasing role in polymer characterization [26], however bias problems concerning ionization efficiency and charge state in dependence on molar mass are well known [27, 28]. In order to analyze a complex sample of oligosaccharides, accurate measurement of relative intensities is required as well. For a small m/z range, relative quantification is possible as it was shown for perdeuteromethylated MC oligomers [8]. For a decamer (degree of polymerization (DP)10), for example, an m/z range up to 90 Da is evaluated since theoretically 0 to 30 Me or Me-d 3 groups per decamer are possible with an m/z difference between two signals of 3 Da.
In HPMC on the other hand, the increase in mass for a single HP substituent is 58 so that, depending on the average MSHP and the DP, the required m/z range for an oligomer can theoretically exceed 750 Da (for DP7, n(HP) = 0–13).
In this work, we studied the effect of selected measurement parameters on the determination of the HP and Me substitution pattern in HPMC using direct sample infusion as well as HPLC-ESI-MS. Moreover, the substituent pattern of HP and Me was evaluated.
Results and discussion
Three HPMCs of comparable DSMe (HPMC1, 1.45; HPMC2, 1.29; and HPMC3, 1.36) but increasing MSHP (HPMC1, 0.28; HPMC2, 0.46; and HPMC3, 0.84) were used for the study. Production of the samples was performed with simultaneous addition of methylation and hydroxypropylation agents. Detailed monomer data on the substituent pattern with respect to position were obtained by gas–liquid chromatography (GLC). GLC in combination with MS allows the assignment of number and position of substituents and thus a differentiated finger print of the sample [29]. However, the exponential increase of possible regio- and diastereoisomers formed with increasing MSHP causes preferably the higher substituted constituents to escape detection. Since the monomer data are the basis for later evaluating the substituent distribution along the polymer chain, their accuracy is of high importance. Therefore, the degree of hydroxypropylation was additionally determined by HPLC-UV after permethylation, total depolymerization and labeling of the monomers with m-aminobenzoic acid (mABA). In contrast to GLC, only slight separation of positional isomers is observed for the HPLC method applied here, enabling the group determination of glucose residues with a certain number of HP groups more sensitively. These molar portions are exactly what is required for the calculation of a reference model for HP distribution in oligomeric sequences of the cellulose chain. The molar fractions of non-, mono-, di- and tri-substituted monomers (c i ) with respect to Me and HP groups from GLC and HPLC analysis are listed in Table 1. More detailed information on the monomer composition with respect to substitution patterns are listed in Table S1 in the Electronic supplementary material (ESM) as well as an example for the calculation of a random distribution of the various glucosyl units in the oligomers (Tables S4 and S5 in the ESM).
Sample preparation
All three HPMC samples were permethylated (for HP pattern analysis) and perdeuteromethylated (for Me pattern analysis) with NaOH/MeI-(-d 3) in DMSO [30]. From permethylated instead of perdeuteromethylated samples, the HP pattern can be determined with higher sensitivity since the intensity of a certain number (n) of HP is not split into several signals (Scheme 1). However, for determination of the Me distribution, the deuteromethylated samples are still required.

ESI-IT mass spectra (negative mode) from HPMC oligomers after permethylation or perdeuteromethylation, partial hydrolysis, and labeling with mABA obtained by means of direct infusion
The peralkylated samples were partially hydrolyzed using acid catalyzed hydrolysis with trifluoroacetic acid (TFA). In order to obtain reliable information on all DPs, partial hydrolysis needs to proceed randomly (without preferred cleavage of a certain substitution pattern). If this requirement is not fulfilled the average MSHP would vary in dependence on hydrolysis time. Subsequently, reducing ends were reductively aminated with mABA [8, 19]. The acidic label enables measurement of the deprotonated form in the negative ion mode. Thus, ionization becomes independent of ion complexation and consequently the labeling strategy is promising to achieve quantitative results. The samples were subjected to ESI-MS (negative mode) analysis and the influence of various parameters was tested.
ESI-MS analysis
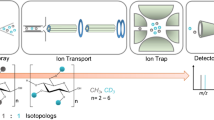
The instrument used for all measurements is a Bruker HCT Ultra ETDII equipped with an IT and an ESI source (Fig. 1).

Setup of the ESI source IT mass spectrometer (modified reproduction with kind permission of Bruker Daltonics)
Sample infusion can be carried out directly by means of a syringe pump or by coupling the mass spectrometer with HPLC. The advantage of direct infusion is the high sample throughput (although by hand) and the possibility to change the measurement parameters during the operation and thus directly observe their effect. Also, the choice of solvents or solvent additives is more flexible than with LC-MS. However, simultaneous infusion of the entire mixture of analytes contained in the sample causes competition of these compounds in all steps of ESI and ion transfer and might favor discrimination effects among the analytes. On the other hand, HPLC separation requires a gradient eluent system; hence, the solvent composition varies with retention time. To study the effect of specific measurement conditions, spectra were first acquired using direct infusion.
For method development, only the permethylated HPMCs were used to determine the HP pattern. This is the larger challenge since the data for one oligomer fraction of a certain DP are extended to an m/z range of several hundred Da due to the mass difference of 58 Da for each HP. Relative ion intensities need to be representative over this entire mass range. In addition to the mass differences, chemical properties of analytes with different substituent pattern, or degree of HP substitution respectively, also influence the electrophoretic mobility and surface activity of the compounds [23, 31].
Also critical for a reliable method are the varying portions with respect to DP depending on the time of hydrolysis. DP1 is always present at highest concentration while concentration is exponentially decreasing with DP [8]. Moreover, within each DP, the amount of oligomers with different number of HP varies. Overall, method development for quantitative evaluation of the HP and Me distribution in HPMC oligomers by ESI-IT-MS needs thorough investigation and the results of our research in this field are presented in the following sections.
Data evaluation
For evaluation of MS data obtained by direct infusion, the intensity of the particular first isotope signal (noise corrected) was used. Based on this, the intensities of the second, third, and fourth isotope signal (i.e., of M + 1, M + 2, and M + 3) were calculated and summed up for evaluation of the relative molar composition of a particular DP.
For perdeuteromethylated HPMC, extracted ion chromatograms (EICs) were generated for oligomers of a certain DP and n(HP). From EICs, an average mass spectrum is generated and the signal intensities are evaluated as described for direct infusion. An example on how calculations were performed in detail is outlined in Table S3 in the ESM.
Variation of ESI-IT-MS measurement parameters
Different parameters of the quantitative oligomer analysis were studied. The influence of degree of depolymerization, solvent composition, and different voltages for the ion transportation unit, respectively trap drive, direct current of the second octopole (Oct2 DC), and OctRF (Fig. 1) are presented.
As a control for the applicability of the measurement parameters used, the MSHP values were calculated from the experimental data for all DPs. Consistency of all MSHP is an important control parameter for a correct analysis because independent of the chain length, the MSHP must match the average value, otherwise data are not representative and indicate bias. In case consistent MSHP values are obtained, the substituent distribution is judged by comparison with the calculated random distribution for the given monomer composition. If so, deviations from this reference model give information on heterogeneities in the sample and their characteristics.
In order to control the hydrolysis time and possible variations of the data, HPMC1 was hydrolyzed for 20, 25, 30, and 40 min to obtain sufficient oligomeric material in the range of DP2–DP7. The DP distribution after different hydrolysis times (almost) represents a most probable distribution if the chain length has no influence on degradation. With increasing hydrolysis time, the amount of monomers increases exponentially while concentration of larger oligosaccharides decreases. All samples obtained from these hydrolysis studies were analyzed under the same conditions by direct infusion ESI-IT-MS. However, the obtained MSHP values do not change significantly with hydrolysis time (see Fig. S1 in the ESM). Thus, the results are independent of the degree of depolymerization.
Whether there is any effect of the ratio of acetonitrile (ACN) and water in the solvent was also tested by syringe infusion. This is interesting with respect to the HPLC-MS studies since gradient elution is required and thus analytes elute under different solvent conditions. Different polarities of the solvent or solvent mixture used could cause a shift in ionization behavior during the electrospray process. The electrophoretic mobility, surface activity, and acidity/basicity of analytes play an important role in ionization especially in analyte mixtures [23, 31]. The higher the surface activity the higher the ion yields. In case of the oligomers from HPMC, a large number of analytes varying in size, polarity, and surface activity is present at the same time, which might be affected to various extents by changing solvent composition.
In order to see if any solvent effects can be expected during HPLC-ESI-MS, HPMC1 was dissolved in ACN/water with different ratios in a range from 40 to 70 % ACN which is the range within which HPMC oligomers elute in gradient HPLC; 1 % glacial acetic acid was added as applied for HPLC-MS. Samples were measured by direct infusion under identical conditions, and the MSHP values obtained for DP2–DP7 were compared. The differences are marginal so that an influence of the ACN/water ratio can be neglected (see Fig. S1 in the ESM).
From the described experiments, it was obvious that the MSHP is not consistent for all DPs. For DP2–DP5, it results in values around 0.3 while for DP7 it is almost 0.2. These strong differences show that measurement parameters (Table 2) used here were not suitable for quantitative evaluation. The spectra obtained from these experiments (not shown here) show low intensity in a higher m/z range which causes underestimation of DP6 and DP7 containing higher number of HP substituents. The detected ratio of analytes does not reflect the real composition in the sample as a result of inappropriate measurement setting.
Parameters responsible for ion formation and ion transportation have a strong effect on the overall ion intensity [24]. With the instrument used in this study, the high voltage of the capillary (HV capillary), the Oct2 DC, and the trap drive were found to have the strongest influence (Fig. 1). The influence of the Skimmer voltage and the lenses 1 and 2 is minor and is not changed here.
The capillary voltage is the potential between the sprayer needle and the capillary and is important for the formation of a good electrospray. The setting for this voltage depends on the solvent that is used. For the mixture of water and ACN that was used in these experiments, the HV capillary was kept at 3.5–4.5 kV to yield a good spray. The trap drive value changes the RF field strength in the IT while higher fields are favorable for higher m/z ions. Voltages of the octopoles influence the focusing of the ions. These parameters were varied and the effect on the resulting spectra was studied.
The following results were obtained by syringe pump infusion with HPMC2 (hydrolyzed for 25 min) and ACN/H2O/NH3 (50/50/0.3, v/v/v) as solvent. Ammonia was added to enhance deprotonation of the mABA tag and improving the signal-to-noise ratio.
An increase of the trap drive leads to a shift of intensities favoring ions with higher m/z (Fig. 2a–d). Ions of lower molecular weight (m/z = 500–700, DP2–3) show constantly high intensities because of their high concentration in the sample, but between m/z = 800 and 1,500, there appears to be a “gap” of intensity, meaning that the parameters are not suitable for a sensitive measurement in that range. Hence, the capacity of the IT for compounds of high molecular weight increases but only in a limited range.

ESI-IT mass spectra (negative mode, [M−H]−) and experimentally determined MSHP values of labeled HPMC2 oligomers (MSHP, 0.46), measured with varying trap drive (A–D) and varying Oct2 DC and OctRF amplitude (E–H). For more detailed settings, see Table 2
Corresponding to this inconsistent intensity distribution, MSHP values were inaccurate for DP2–DP7. The MSHP for DP6 and DP7 become too high with increasing trap drive since the signals for high n(HP) are overestimated under these conditions.
When trap drive is kept constant the parameters of the ion transportation unit have to be optimized. The two octopoles are responsible for the focusing and transportation of the ions towards the IT. The direct current (DC) of both octopoles as well as the radio frequency (RF) can be varied. The Oct1 DC has a default value of −8 V and was not changed in this study. However, using a medium trap drive value (140.9), increase of the Oct2 DC and OctRF amplitude lead to reduction of the intensity-gap in the mass range of 1,000–1,500 m/z (Fig. 2e–h). With an Oct2 DC of −5.10 to −5.65 V and OctRF amplitude between 233.5 and 254.4 Vpp, good conformity of MSHP values can be achieved for DP3–DP6. The deviations for DP2 (higher than reference) and DP7 (lower than reference) are discussed in connection with the resulting HP distributions in the following paragraph.
Results for the HP substitution pattern
For the determination of the HP pattern, syringe pump infusion with ACN/H2O/NH3 as solvent and optimized ion transportation settings (Table 2, HP pattern) were finally applied. The MSHP values obtained for all three HPMC together with the corresponding HP distribution are shown in Fig. 3. These relative distributions are plotted as a function of the MSHP to enable an easy data comparison among the DPs and the samples respectively.

Comparison of calculated random (filled squares) and experimental (empty squares) HP distribution for DP2–DP7 of HPMC1 (MSHP, 0.28), HPMC2 (MSHP, 0.46), and HPMC3 (MSHP, 0.84) obtained by ESI-IT-MS (direct infusion, negative mode) after permethylation, partial hydrolysis, and reductive amination with mABA. Average MSHP values are an average of three measurements (for standard deviations, see Table S2 in the ESM)
HPMC1 (MSHP, 0.28) shows very little deviation from the random model. For all DPs, an average MSHP of 0.29 was determined with the exception of DP2. MSHP of DP2 is significantly overestimated in all samples, and the distribution shows a too low amount of nonhydroxypropylated dimers. The reason for this can probably be found in the measurement parameters which slightly suppress the ion yield of this particular m/z = 546. Since this effect is found in all samples, DP2 is not discussed for HPMC2 and HPMC3.
With 0.47–0.48, the MSHP values found for the oligomeric domains of HPMC2 (MSHP, 0.46), are very close to the expected reference data excepting DP7. Obviously, the intensities in the higher mass range are already too low for satisfactory quantitative evaluation for DP7. Since HPMC1 has a lower MSHP, this problem does not arise. Having a closer look on the HP distribution in HPMC2 (DP3–DP6), a slightly broader curve is obtained from the experimental data compared with the calculated ones. The portion of n(HP) = 0 as well as the amount of higher substituted oligomers is enhanced, compensated by the lower contribution at the average MSHP. This indicates a slightly more heterogeneous compared with a random HP distribution.
HPMC3 shows stronger deviations for MSHP values as well as for the HP distributions. The MSHP values are higher than the reference value (MSHP, 0.84).
This is quite possible since reference values obtained by HPLC-UV might be too low when tiny amounts of higher substituted constituents escape detection. However, the shape of the HP distribution curves are consistent with the random distributions, but only slightly shifted to a higher MS.
In conclusion quantitative ESI-IT-MS with direct sample infusion can be successfully applied to permethylated HPMCs in a MSHP range at least up to about 0.5 for the determination of the HP pattern when optimized measurement parameters are used.
Results for the Me substitution pattern
The determination of the Me pattern in HPMC gives insight into another structural level of the substrate. For each n(HP), a subpattern for Me is observed and more differentiated analysis is possible.
However, direct infusion is no longer applicable due to interference of compounds with nearby m/z of two consecutive DP with different numbers of Me and HP groups. Using LC-MS, these interfering analytes are separated, and mass spectra can be evaluated individually using EICs. The perdeuteromethylated samples of HPMC1–3 were analyzed applying optimized parameters as described for the HP pattern. Compared with direct infusion measurement conditions differed only with respect to solvent and nebulizing settings (Table 2, Me pattern). LC-MS was carried out on a reversed phase (RP18) column with gradient elution using ACN and water as mobile phase each containing 1 % of acetic acid instead of NH3. The acetic acid suppresses deprotonation of the mABA label on the column and improves chromatographic separation. In spite of this acid addition, dissociation of mABA in the negative mode ESI-MS is not suppressed. The flow rate used for LC-MS was 0.2 mL/min, thus dry gas temperature, nebulizer pressure and dry gas flow were enhanced.
The resulting average DSMe values for DP2–5 are shown in Fig. 4 as well as the experimentally determined Me distribution (weighted average). Within one oligomer fraction, Me distributions can be evaluated depending on the number of HP groups. These particular Me patterns were summed up weighting their individual contribution. These contributions were taken from the analysis of the HP pattern after permethylation. The obtained DSMe values should be consistent throughout all DPs and fit the data from monomer analysis.

Comparison of calculated random (filled squares) and experimental (empty squares) Me distribution in DP2–DP5 of HPMC1 (DSMe, 1.45), HPMC2 (DSMe, 1.29), and HPMC3 (DSMe, 1.36) obtained by ESI-IT-MS (LC-MS, negative mode) after perdeuteromethylation, partial hydrolysis, and reductive amination with mABA
For HPMC1 and HPMC2 the average DSMe values fulfill this requirement within experimental error. The increase of the weighted averaged DSMe for HEMC3 is probably due to the overall low intensity of the relatively broad Me distribution. For HPMC2 and HPMC3 DSMe obtained from oligomer evaluation is slightly lower than determined from GLC analysis. However, the comparison of the experimentally determined Me pattern with the calculated random distribution (Fig. 4) unambiguously shows a distorted profile with a more linear slope at the low DS side. This indicates a DS gradient in the material, since the more linear shape of the left flank is the envelope of a number of sub-patterns of different DS [32].
The Me patterns can be further differentiated with respect to the number of HP groups. Looking at the correlation of the DSMe and MSHP (Fig. 5) for all DPs of HPMC1, it is obvious that DSMe steadily decreases with increasing MSHP while the total DS (DSMe + MSHP) increases. This is plausible since, as a result of a random Me and HP distribution, the cellulose chains exhibit regions with slight differences in substitution, thus higher MSHP means higher overall substitution. The decrease of DSMe with increasing number of HP (Fig. 5) in the oligomers can be explained with the competition of MeCl and Me oxirane for the deprotonated OH groups in the glucose units during the simultaneous manufacturing process of HPMC. Although the ring opening of Me oxirane causes in situ formation of a new deprotonated OH group, this alkoxy group shows less reactivity for methylation than the OH groups of the sugar ring. In aqueous media it is probably protonated quickly and therefore loses reactivity. The low reactivity of HP OH can already be observed from monomer data (see Table S1 in the ESM) showing that only 10-15 % of HP-OH, but 42-48 % of backbone-OH are methylated. This trend has already been observed in earlier work [17]. For hydroxyethyl groups in HEMC on the other hand an increase of DSMe with increasing n(HE) is observed [33]. Thus HE acts as a spacer, HP as a competitor for Me.

Variation of the DSMe and total DS (DSMe + MSHP) with increasing MSHP in DP2–DP5 of HPMC1 (DSMe, 1.45), HPMC2 (DSMe, 1.29), and HPMC3 (DSMe, 1.36) obtained by HPLC-MS (negative mode)
HPMC2 and HPMC3 show a significantly lower DSMe with respect to the average DSMe at very low MSHP values (0–0.4) (Fig. 5). Deviation is also DP-dependent. Thus the curves do no longer start at the same DSMe for MSHP = 0. This could be caused by the existence of poorly activated (deprotonated) areas in the cellulose bulk material during the manufacturing process of the HPMCs. These areas have a low degree of HP and Me substitution as well, and they are more easily observed for larger oligomers. Differentiation of Me patterns with respect to n(HP) is shown for DP2–5 of all HPMCs in Fig. 6.

For HPMC1, the Me subpatterns for 0, 1, 2, 3, and 4 HP groups are stepwise shifted from higher to lower DSMe while maintaining the general shape of the profile. This behavior is expected since a certain position of the cellulose can only be occupied by a Me or by a HP residue, because only 10–12 % of the HP groups are methylated as mentioned above.
The shape of the experimentally found Me patterns for HPMC2 and HPMC3 are obviously distorted (see Fig. 4). From the Me subpatterns of these samples (Fig. 6), it is evident that the low substituted domains with no HP show a broad distorted profile typical for strongly heterogeneous substitution. This heterogeneity is—even if less pronounced—observed for the oligomers with n(HP) = 1 and becomes less and less visible with increasing number of HP groups.
One has to keep in mind that the portion with n(HP) = 0 only accounts for a relatively small percentage of the sample which decreases with increasing DP (Table 3).
However, these deviations from a random substituent distribution also explain the slight deviations found for the HP distribution in HPMC2. Often a small amount of poorly activated and thus poorly substituted material strongly influences the properties of the bulk material [9]. By comprehensive evaluation of the analytical data, it turns out that HP can be used as a probe for internal fractionation according to DSMe. Differentiation according to n(HP) allows the extraction of Me subpatterns and thus strongly enhances the sensitivity for the detection of deviations from randomness in the material.
Conclusions
Three HPMCs with DSMe of 1.45, 1.29, and 1.36 and MSHP of 0.28, 0.46, and 0.84 have been analyzed with respect to their Me and HP distribution along the cellulose backbone by means of ESI-IT-MS. The sample preparation procedure includes per(deutero)methylation, partial hydrolysis, and reductive amination with mABA. In order to obtain reliable information on both types of substituent distributions, a selected set of method parameters was varied and the effect on the mass spectra and data evaluation was studied. The time of hydrolysis as well as the ratio of ACN and water for direct sample infusion had no influence in the ionization efficiency and the relative intensities of the resulting mass spectra.
Variation of ion transportation parameters like direct current of the octopoles and the trap drive of the IT on the other hand, show pronounced effects on the mass spectra. Consistent MSHP values for DP3–DP6 of HPMC1 (MSHP = 0.28) and HPMC2 (MSHP = 0.46) were obtained using optimized measurement conditions and direct infusion with a syringe pump, while for HPMC3 the average MSHP was not met, but qualitative evaluation of the profile was possible. The Me distribution was successfully determined by HPLC-ESI-MS employing perdeuteromethylation. While HPMC1 shows a random distribution of both, Me and HP, HPMC2 and HPMC3 showed significant deviation from random profiles. These deviations could be detected even more pronounced by more detailed evaluation of the analytical data, using HP as a probe to look into Me sub-patterns.
Experimental part
Materials
All HPMCs were prepared by Dow Wolff Cellulosics. For their preparation ground cellulose flock was added to a 5-L autoclave. After purging the autoclave twice with nitrogen, caustic soda (NaOH) was added. Then, Me chloride, propylene oxide, and dimethyl ether were added. The mixture was heated to 80 °C and the reaction was completed. The amounts of reactants were adjusted depending on the targeted DSMe and MSHP. The crude cellulose ether was then cooled down, vacuum was pulled and then neutralized with formic acid and washed free of chloride with hot water cooled to room temperature (r.t.) and dried at 55 °C in an air-swept dryer.
Solvents used for sample preparation for ESI-MS were of HPLC gradient grade or LC-MS grade. All reagents for reductive amination were obtained from Sigma-Aldrich (St. Louis, USA).
ESI-IT-MS
ESI-IT mass spectra were recorded on a HCT Ultra ETDII (Bruker Daltonics, Bremen, Germany), equipped with an IT. Mass spectra evaluation was performed with Data Analysis (Bruker Daltonics, Bremen, Germany).
For direct infusion, the analyte solutions (∽0.02 mg/mL) were infused with a syringe pump at a flow rate of 200 μL/h. As solvents methanol, ACN, water (Nanopure®), and mixtures of these were used with addition of aqueous ammonia solution or glacial acetic acid. Each spectrum is an average of 200 scans. Nitrogen was used as dry gas and as nebulizer gas. Detailed measurement parameters are listed in Table 2. LC-MS separation was carried out on an Agilent HPLC system consisting of a binary pump (1100 Series) and a DAD (G1312A). A RP18-column (Phenomenex, Kinetex, 2.6 μm, 100 × 2.1 mm) was used with H2O/HOAc (99/1, v/v; A) and ACN/HOAc (99/1, v/v; B) in a linear gradient system (0 min, 90 % A and 30 min, 20 % A) and a flow rate of 0.2 mL/min. Sample concentration was 0.2 mg/mL and injection volume was set to 10 μL.
Sample preparation
Alkylation
Per(deutero)methylation was performed with pulverized NaOH and MeI-(−d 3) in DMSO at r.t.. Prior to the reaction the samples and glass ware were dried in high vacuum. Samples were dissolved in DMSO (50 mg/5 mL) and freshly powedered NaOH (6 mol-eq./anhydroglucose unit (AGU)) was added. After 15 min of stirring, MeI-(d 3) (4 mol-eq./AGU) was added and the mixture was stirred for 12 h.
Sample clean-up was performed by dialysis (MWCO of 14 kDa) of the reaction mixture against water. Afterwards the sample was lyophilized and completeness of the reaction was controlled by ATR-IR spectroscopy.
Partial hydrolysis
In a 1-mL V-vial ca. 2 mg of fully alkylated HPMCs (∽10 μmol AGU ) were dispersed in 925 μL acetone/water (10:8.5 v/v). After 2 h at r.t. 75 μL concentrated TFA were added. The mixture was kept at 120 °C for 25 min. For the hydrolysis time study, 20, 30, and 45 min were also used.
After cooling, the sample was co-evaporated with toluene to dryness under a stream of nitrogen in order to remove the solvents and TFA.
Labeling with mABA
To the residue obtained after partial hydrolysis was dissolved in 0.5 mL methanol and 0.3 mL of mABA-solution (containing 2.7 mg; 20 μmol) as well as 0.15 mL glacial acetic acid were added. The mixture was heated to 40 °C for 30 min. Afterwards reduction was performed by adding 50 μL of a 2-picoline borane solution in methanol (containing 2.1 mg; 20 μmol) and holding the mixture at 40 °C for 45 min. After cooling to r.t. the solvent and acid were removed in a stream of nitrogen and the residue was dissolved in 1 mL MeOH (stock solution, 2 mg/mL).
Abbreviations
- ACN:
-
Acetonitrile
- AGU:
-
Anhydroglucose unit
- DP:
-
Degree of polymerization
- DSMe :
-
Degree of methyl substitution
- EIC:
-
Extracted ion chromatogram
- ESI:
-
Electrospray ionization
- GLC:
-
Gas–liquid chromatography
- HP:
-
Hydroxypropyl
- HPLC:
-
High-performance liquid chromatography
- HPMC:
-
Hydroxypropyl methylcellulose
- IT:
-
Ion trap
- mABA:
-
m-aminobenzoic acid
- Me:
-
Methyl
- MS:
-
Mass spectrometry
- MSHP :
-
Molar degree of hydroxypropyl substitution
- r.t.:
-
Room temperature
References
Thielking H, Schmidt M (2006) Cellulose ethers. In: Ullmann's Encyclopedia of Industrial Chemistry. Wiley, New York
Siepmann J, Kranz H, Bodmeier R, Peppas NA (1999) HPMC-matrices for controlled drug delivery: a new model combining diffusion, swelling, and dissolution mechanisms and predicting the release kinetics. Pharm Res 16:1748–1756
Haque A, Richardson RK, Morris ER, Gidley MJ, Caswell DC (1993) Thermogelation of methylcellulose. Part II: effect of hydroxypropyl substituents. Carbohydr Polym 22:175–186
Burchard W (2003) Solubility and solution structure of cellulose derivatives. Cellulose 10:213–225
Mischnick P, Momcilovic D (2010) Chemical structure analysis of starch and cellulose derivatives. Adv Carbohydr Chem Biochem 64:117–210
Viridén A, Wittgren B, Andersson T, Abrahmsén-Alami S, Larsson A (2009) Influence of substitution pattern on solution behavior of hydroxypropyl methylcellulose. Biomacromolecules 10:522–529
Adden R, Müller R, Mischnick P (2006) Fractionation of methyl cellulose according to polarity—a tool to differentiate first and second order heterogeneity of the substituent distribution. Macromol Chem Phys 207:954–965
Cuers J, Unterieser I, Burchard W, Adden R, Rinken M, Mischnick P (2012) Simultaneous determination of substituent patterns in partially acid hydrolyzed O–Me/O–Me-d 3-cellulose and quantification of the obtained oligomers by HPLC-ESI-MS. Carbohydr Res 348:55–63
Mischnick P, Adden R (2008) Fractionation of polysaccharide derivatives and subsequent analysis to differentiate heterogeneities on various hierarchical levels. Macromol Symp 262:1–7
Arisz PW, Kauw HJJ, Boon JJ (1995) Substituent distribution along the cellulose backbone in O-methylcelluloses using GC and FAB-MS for monomer and oligomer analysis. Carbohydr Res 271:1–14
Mischnick P, Heinrich J, Gohdes M, Wilke O, Rogmann N (2000) Structure analysis of 1,4-glucan derivatives. Macromol Chem Phys 201:1985–1995
Heinrich J, Mischnick P (1996) Rapid method for the determination of the substitution pattern of O-methylated 1,4-glucans by high-pH anion-exchange chromatography with pulsed amperometric detection. J Chromatogr A 749:41–45
Voiges K, Adden R, Rinken M, Mischnick P (2012) Critical re-investigation of the alditol acetate method for analysis of substituent distribution in methyl cellulose. Cellulose 19:993–1004
Lazik W, Heinze T, Pfeiffer K, Albrecht G, Mischnick P (2002) Starch derivatives of a high degree of functionalization. VI. Multistep carboxymethylation. J Appl Polym Sci 86:743–752
Mischnick P (2012) Mass spectrometric characterization of oligo- and polysaccharides and their derivatives. Adv Polym Sci 248:105–174
Lamari FN, Kuhn R, Karamanos NK (2003) Derivatization of carbohydrates for chromatographic, electrophoretic and mass spectrometric structure analysis. J Chromatogr B 793:15–36
Adden R, Müller R, Mischnick P (2006) Analysis of the substituent distribution in the glucosyl units and along the polymer chain of hydroxypropylmethyl celluloses and statistical evaluation. Cellulose 13:459–476
Unterieser I, Cuers J, Voiges K, Enebro J, Mischnick P (2011) Quantitative aspects in electrospray ionization ion trap and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of malto-oligosaccharides. Rapid Commun Mass Spectrom 25:2201–2208
Unterieser I, Mischnick P (2011) Labeling of oligosaccharides for quantitative mass spectrometry. Carbohydr Res 346:68–75
Schmidt A, Karas M, Dülcks T (2003) Effect of different solution flow rates on analyte ion signals in nano-ESI MS, or: when does ESI turn into nano-ESI? J Am Soc Mass Spectrom 14:492–500
Tang L, Kebarle P (1993) Dependence of ion intensity in electrospray mass spectrometry on the concentration of the analytes in the electrosprayed solution. Anal Chem 65:3654–3667
Schriemer DC, Li L (1997) Mass discrimination in the analysis of polydisperse polymers by MALDI time-of-flight mass spectrometry. 2. Instrumental issues. Anal Chem 69:4176–4183
Kebarle P, Verkerk UH (2009) Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom Rev 28:898–917
Kruve A, Herodes K, Leito I (2010) Optimization of electrospray interface and quadrupole ion trap mass spectrometer parameters in pesticide liquid chromatography/electrospray ionization mass spectrometry analysis. Rapid Commun Mass Spectrom 24:919–926
Titato GM, Bicudo RC, Lanças FM (2007) Optimization of the ESI and APCI experimental variables for the LC/MS determination of s-triazines, methylcarbamates, organophosphorous, benzimidazoles, carboxamide and phenylurea compounds in orange samples. J Mass Spectrom 42:1348–1357
Murgasova R, Hercules D (2002) Polymer characterization by combining liquid chromatography with MALDI and ESI mass spectrometry. Anal Bioanal Chem 373:481–489
Gruendling T, Guilhaus M, Barner-Kowollik C (2008) Quantitative LC-MS of polymers: determining accurate molecular weight distributions by combined size exclusion chromatography and electrospray mass spectrometry with maximum entropy data processing. Anal Chem 80:6915–6927
Byrd HCM, McEwen CN (2000) The limitations of MALDI-TOF mass spectrometry in the analysis of wide polydisperse polymers. Anal Chem 72:4568–4576
Mischnick P (1989) Determination of the patterns of substitution of hydroxyethyl- and hydroxypropyl-cyclomaltoheptaoses. Carbohydr Res 192:233–241
Ciucanu I, Kerek F (1984) A simple and rapid method for the permethylation of carbohydrates. Carbohydr Res 131:209–217
Zhou S, Cook KD (2001) A mechanistic study of electrospray mass spectrometry: Charge gradients within electrospray droplets and their influence on ion response. J Am Soc Mass Spectrom 12:206–214
Mischnick P, Kühn G (1996) Model studies on methyl amyloses: correlation between reaction conditions and primary structure. Carbohydr Res 290:199–207
Mischnick P, Unterieser I, Voiges K, Cuers J, Rinken M, Adden R (2013) A new method for the analysis of the substitution pattern of hydroxyethyl(methyl)-celluloses along the polysaccharide chain. Macromol Chem Phys (in press)
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Separation and Characterization of Natural and Synthetic Macromolecules with guest editors Albena Lederer and Peter J. Schoenmakers.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 2057 kb)
Rights and permissions
About this article
Cite this article
Cuers, J., Rinken, M., Adden, R. et al. Critical investigation of the substituent distribution in the polymer chains of hydroxypropyl methylcelluloses by (LC-)ESI-MS. Anal Bioanal Chem 405, 9021–9032 (2013). https://doi.org/10.1007/s00216-013-7065-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-7065-0