
Abstract
Carbon isotope ratio (CIR) analysis has been routinely and successfully applied to doping control analysis for many years to uncover the misuse of endogenous steroids such as testosterone. Over the years, several challenges and limitations of this approach became apparent, e.g., the influence of inadequate chromatographic separation on CIR values or the emergence of steroid preparations comprising identical CIRs as endogenous steroids. While the latter has been addressed recently by the implementation of hydrogen isotope ratios (HIR), an improved sample preparation for CIR avoiding co-eluting compounds is presented herein together with newly established reference values of those endogenous steroids being relevant for doping controls. From the fraction of glucuronidated steroids 5β-pregnane-3α,20α-diol, 5α-androst-16-en-3α-ol, 3α-Hydroxy-5β-androstane-11,17-dione, 3α-hydroxy-5α-androstan-17-one (ANDRO), 3α-hydroxy-5β-androstan-17-one (ETIO), 3β-hydroxy-androst-5-en-17-one (DHEA), 5α- and 5β-androstane-3α,17β-diol (5aDIOL and 5bDIOL), 17β-hydroxy-androst-4-en-3-one and 17α-hydroxy-androst-4-en-3-one were included. In addition, sulfate conjugates of ANDRO, ETIO, DHEA, 3β-hydroxy-5α-androstan-17-one plus 17α- and androst-5-ene-3β,17β-diol were considered and analyzed after acidic solvolysis. The results obtained for the reference population encompassing n = 67 males and females confirmed earlier findings regarding factors influencing endogenous CIR. Variations in sample preparation influenced CIR measurements especially for 5aDIOL and 5bDIOL, the most valuable steroidal analytes for the detection of testosterone misuse. Earlier investigations on the HIR of the same reference population enabled the evaluation of combined measurements of CIR and HIR and its usefulness regarding both steroid metabolism studies and doping control analysis. The combination of both stable isotopes would allow for lower reference limits providing the same statistical power and certainty to distinguish between the endo- or exogenous origin of a urinary steroid.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Within the last years, the number of investigations on carbon isotope ratios (CIR) of endogenous urinary steroids in the context of sports drug testing has considerably increased [1-14]. Regarding absolute values expressed as δ13CVPDB against the international standard Vienna Pee Dee Belemnite (VPDB) based on Eq. 1, large differences were detected between different investigated populations [15].
This finding is not surprising as δ13C-values of steroids are mainly influenced by the CIR of the ingested diet resulting in significantly different values for athletes from different geographical origins [6, 7]. In sports drug testing, absolute δ13C-values of urinary steroids are not the first choice as many cases of steroid misuse might go undetected especially if only a small amount of steroid was administered [5]. Differences between an endogenous reference compound (ERC) such as pregnanediol (PD) and a target compound (TC) like testosterone (TESTO) expressed as Δ-values based on Eq. 2 have been of greater utility.
The World Anti-Doping Agency (WADA) has defined one threshold at 3 ‰ for all possible Δ-values [16]. As isotope ratios are influenced by fractionation processes, not every urinary steroid possesses the same isotopic composition despite the fact that they are all derived from the same precursor molecule cholesterol [4-6, 17]. Additionally, different isotope ratios were found at different steroid production sites of the human body [18]. So Δ-values established by means of reference population-based studies seem to be more promising in detecting steroid misuse. Taking into account the found differences between different steroids helps to avoid false positive findings as the defined reference limit reflects the found natural occurring differences in-between the steroids. Furthermore, they cover both the intra- and inter-individual variations as well as all measurement uncertainties and can readily be applied to routine doping control samples after being established properly [19, 20].
During a former comprehensive investigation on CIR of urinary steroids investigating ten different compounds, it was hypothesized that insufficient chromatographic resolution might result in influenced δ13C-values [5]. Within this study, sample preparation was adapted and improved to yield completely separated compounds which enabled measurements of almost each steroid individually. Particular attention was paid to the isolation of 5α- and 5β-steroids.
Implemented into this approach were 3α-hydroxy-5β-androstane-11,17-dione (11KETO), 5β-pregnane-3α,20α-diol (PD), 5α-androst-16-en-3α-ol (16EN), 17β-hydroxy-androst-4-en-3-one (TESTO), 17α-hydroxy-androst-4-en-3-one (EPIT), 3β-hydroxy-androst-5-en-17-one (DHEA), 5α-androstane-3α,17β-diol (5aDIOL), 5β-androstane-3α,17β-diol (5bDIOL), 3α-hydroxy-5α-androstan-17-one (ANDRO) and 3α-hydroxy-5β-androstan-17-one (ETIO) excreted into urine as glucuronide conjugates. Moreover steroid sulfates of ETIO, ANDRO, DHEA and 3β-hydroxy-5α-androstan-17-one (EPIA), androst-5-ene-3β,17β-diol (5EN17b) and androst-5-ene-3β,17α-diol (5EN17a) were included.
As influences on CIR by changing the sample preparation method were expected, investigations on a new reference population encompassing 67 individuals became necessary and allowed for comparison studies to formerly established reference-based thresholds. Additionally, the issue of instrument linearity has been addressed in a separate experiment as no sound data on the amount-dependent behavior of steroids regarding mainly the combustion process has been presented in literature so far.
As the above-mentioned reference population was also investigated regarding hydrogen isotope ratios (HIR) of urinary steroids [13], the feasibility of combining both stable isotopes for enhanced drug testing and metabolic studies was investigated, too. In this context, special attention will be paid to an administration study with TESTO-undecanoate administered orally where both stable isotopes were assayed.
Experimental
Chemicals and steroids
BakerbondTM SPE Octadecyl columns (6 mL, 500 mg) were purchased from J.T. Baker (Deventer, Netherlands). tert-Butyl methyl ether (TBME) was obtained from Acros (Geel, Belgium), methanol from Merck (Darmstadt, Germany) and acetonitrile from Biosolve (Valkensward, Netherlands). Pyridine and acetic anhydride were from Sigma-Aldrich (Buchs, Switzerland) and β-glucuronidase from Escherichia coli from Roche Diagnostics GmbH (Mannheim, Germany). All solvents and reagents were of analytical grade.
Steroid reference material 5aDIOL, 5bDIOL, 16EN, 11KETO, 5EN17b, 5EN17a, EPIA, and 3β-hydroxy-5α-androstane (RSTD) were supplied by Steraloids (Newport, RI, USA). ANDRO, ETIO, PD, DHEA, EPIT, TESTO and TESTO-undecanoate were supplied by Sigma-Aldrich (Steinheim, Germany). For calibration of the carbon dioxide tank gas (Carbagas, Gümlingen, Switzerland) a n-alkane mixture #0452.3 from Chiron (Trondheim, Norway) was used together with the USADA mixture 33-1 provided by Cornell University (Ithaca, NY, USA) containing acetylated ANDRO, 11KETO and RSTD plus underivatized 5α-cholestane (CHOL) [21]. Another secondary reference material was purchased from A. Schimmelmann (Indiana University, IN, USA), namely the n-alkane mixture C3 containing heptadecane, nonadecane, heneicosane, tricosane, and pentacosane which was used in the linearity experiments.
Reference population
Urine specimens from 38 males and 29 females, aged from 20 to 54 years were collected in the forenoon and stored frozen at −20 °C until analysis. All volunteers were healthy, exercised on a regular basis up to 20 h/week and were recruited from students and employees of the German Sports University Cologne (Germany) and the Swiss Laboratory for Doping Analysis (Switzerland). All volunteers were requested to fill in a short questionnaire. The study was approved by the local ethical committee of the German Sport University Cologne, and written consent was given by all participants.
Excretion study
One healthy male volunteer (37 years, 82 kg, 180 cm) administered orally 100 mg of TESTO-undecanoate dissolved in ethanol/water (40/60 v/v). The ethical committee of the canton Vaud (Protocol 155/11) and Swissmedic (reference number 2011DR3149) approved the study, and the participant gave written consent.
Prior to the administration study, the δ13CVPDB and δ2HVSMOW (hydrogen isotope ratios are expressed in parallel to Eq. 1 using Vienna Standard Mean Ocean Water (VSMOW) as international reference standard) values of both TESTO-undecanoate and the unconjugated TESTO were determined. Ester cleavage was accomplished according to the method applied by Cawley et al. [22]
Three negative-control urine samples were collected before drug administration and all spontaneous urine specimens for the following 2 days. Subsequently, only the morning and one evening urine were sampled for 3 days. This protocol resulted in a total number of 21 urines. All specimens were stored frozen until preparation.
GC/MS measurements
Both identification and quantification were conducted as described in detail in the previous publication on HIR of the same reference population [13].
Sample preparation
Also, the extensive sample preparation procedure including several solid phase and liquid–liquid extraction steps was described together with the well-elaborated high performance liquid chromatography (HPLC) clean up procedure [13].
The main difference between this method and the previously published assay afflicted with influenced CIR values [5] was the alteration of the HPLC column from a commonly used C18 column to a XBridgeTM Shield RP18 column purchased from Waters (Baden-Dättwil, Switzerland). This column showed an improved separation power for all steroids differing only in their 5α- or 5β-conformation such as, e.g., ETIO and ANDRO or 5aDIOL and 5bDIOL.
GC/C/IRMS measurements
All CIR were measured on an Agilent 7890 Gas Chromatograph (Waldbronn, Germany) coupled to a Delta V gas isotope ratio mass spectrometer (IRMS) (ThermoFisherScientific, Bremen, Germany) via the GC combustion interface (GCC III, ThermoFisherScientific). Injections were performed in splitless mode at 280 °C with injection volumes ranging from 1 to 3 μL of cyclohexane. The GC column was a J&W Scientific DB-17MS (length 30 m, i.d. 0.25 mm, film thickness 0.25 μm) from Agilent. The initial oven temperature of 70 °C was maintained for 2 min and increased at 30 °C/min up to 270 °C, then at 2 °C/min to 290 °C and then at 30 °C/min up to 300 °C and kept for 3 min. Carrier gas was purified He (purity grade 4.9) with a constant flow of 1.4 mL/min. The combustion furnace was operated at 940 °C. Data acquisition was done using ISODAT® 3.0 software (ThermoFisherScientific).
The HIR measurements and the experiments regarding instrument linearity were all performed on the described set up using a Thermo Deltaplus XL IRMS [13].
Calibration of the tank gas
The apparent δ13CVPDB-value of the carbon dioxide tank gas (−29.4 ‰) was calculated using the mean value of all n-alkanes comprised in the Chiron mixture. The ascertained value for the tank gas was affirmed using the USADA 33-1 steroid mixture to follow the principle of identical treatment of standard and analytes [23].
Negative-control urine
Five spot urine samples from different individuals who declared no use of any prohibited substance or nutritional supplement were pooled and stored frozen at −20 °C in 20 mL portions. This urine was used for both, measurements regarding repeatability and stability over time.
Correction for the acetate moiety
All determined values were corrected for the influence of the acetate moiety as described in literature [5, 13, 24]. All CIR and HIR of urinary steroids reported within this article are for the underivatized steroid.
Method validation
Repeatability of the entire procedure was tested for by six replicate preparations of one negative-control urine and subsequent determination of CIR values. The method’s stability over time was monitored over 8 months by preparing one negative-control urine with each batch of samples processed for the reference population determinations (n = 6). Further steps of validation were not subject of this investigation as the complete method was carefully assessed in earlier studies [5, 11].
Instrument linearity
Within continuous flow IRMS applications linearity, of the mass spectrometer is always crucial as peak heights may vary over a wide range. Especially for CIR determinations in doping control analysis this might constitute a problem as here the variability is pronounced due to large concentration differences between all urinary steroids ranging from low nanogram-per-milliliter to midrange microgram-per-milliliter amounts. Besides, the method was improved to enable separate determinations of nearly each steroid, which allows reconstituting every fraction on its own to reach approximately equal peak height, especially the low concentrated compounds still result in low intensities.
Therefore, the linearity of the GC/C/IRMS apparatus as a whole was systematically investigated by repeated injections at different concentrations of both the n-alkane mixture C3 and the USADA 33-1 steroid mixture. Four subsets of measurements were conducted with 48 injections of both the alkanes and the steroids at eight different concentration levels respectively summing up in 192 measurements for each class of compounds. The measurements were completed within 4 weeks and throughout this interval the linearity of the MS was tested for by standard_on_off tank gas peaks at ten different intensities yielding a MS-linearity <0.03 ‰/V.
Combination of CIR and HIR
Both stable isotopes evaluated individually enable the detection of steroid misuse and present equally valuable approaches as outlined below by means of the TESTO administration study (vide infra). However, both isotope-based analytical methodologies can fail in detecting exogenous steroids as for both parameters pharmaceutical preparation with endogenous signatures were reported [22, 25]. Moreover, CIR of endogenous steroids can be close to those observed with synthetic products as described for athletes from northern Europe [7]. Hence, the possibility of combining threshold Δ-values of CIR and HIR at a lower level of significance (p = 0.05) was investigated together with any correlation between both isotopes which might proscribe the chosen approach.
Results and discussion
Method validation
The results obtained for short- and long-term repeatability are listed in Table 1. Overall, the standard deviations (SD) were found to be similar to those published for this method earlier [5, 11]. Only EPIT is found with slightly higher SD than the other steroids. This phenomenon has already been described in literature [5, 9]. The mean values did not change over time indicating the stability of measurements and excluding the introduction of any bias by sample preparation or isotope analysis.
Instrument linearity
While the linearity of the MS or the ion source, respectively, is tested by standard CO2 tank gas on–off pulses at varying peak heights and, if required, corrected by re-focusing of the ion source, the linearity of the whole GC/C/IRMS system can only be verified by repeated injections of standards at different concentration. To differentiate factors influencing only steroids from general factors also affecting other classes of substances, n-alkanes were investigated in parallel to the USADA steroid mixture. As shown in Fig. 1, differences between both analytes are obvious. While all five n-alkanes show a similarly excellent linearity over the dynamic range of the instrument from 100 up to 11,000 mV peak height (Fig. 1, upper part), the steroids did not feature this homogeneity. CHOL and RSTD are linear down to 20 mV; however, ANDRO and especially 11KETO exhibit a substantial variability of their CIR depending on the peak intensity. Especially one subset of measurements (represented by the lowest values for each steroid) showed deviation up to 4 ‰ from the certified value. After this first set of measurements, the connection between the GC column and the combustion furnace was renewed. This improved the linearity but could not entirely eliminate the effect leaving an offset for 11KETO up to 1.5 ‰. Most probably, this effect does not result from incomplete combustion as the other investigated steroids were not affected but is due to the incomplete transfer from the GC column onto the hot zone of the combustion furnace. Such issues were described earlier and necessitate consideration [26, 27].

Results obtained for the instrument linearity experiment. In the upper part the results for the n-alkane mixture are depicted, the USADA mixture can be found in the lower part. The assignment is described in the caption together with the certified values of each compound given in δ13CVPDB [‰]. The lines represent the best linear fit with the exemption of 11KETO and ANDRO where a logarithmic fit suits better
Additionally, an amount-dependent shift of the IRMS response among the different steroids of the USADA mixture was observed. As shown in Figure S1 (see Electronic Supplementary Material), the ratio of the area under the peak of 11KETO or ANDRO divided by the area of the belonging RSTD varies with the amount of steroid injected on-column. These amount-dependent losses of 11KETO and ANDRO might offer another explanation for their changes in CIR responses. Other effects caused by the GC system such as losses linked to the split/splitless injector might also contribute to the described behavior. Further studies should enable to locate the different factors that contribute to the shift in δ-values.
Nevertheless, this highlights the necessity of adopting the principle of identical treatment to steroids and that calibrations only performed with n-alkanes might not account for system-immanent issues. As a consequence of these findings, possible correlations between the urinary concentrations of 11KETO and ANDRO and the respective δ-values were investigated to identify systematic offsets. No correlations were detected, arguably due to the relatively high concentrations of these steroids and the possibility to adjust the peak height separately for each measured compound. A general systematic offset for any steroid cannot be excluded but would have been implemented into the reference-based thresholds.
Another interesting point is that the SD decreases with peak intensities (see Electronic Supplementary Material Fig. S2). This has sporadically been described for selected steroids (norandrosterone and boldenone) but never investigated carefully [28, 29]. Again, both datasets could be evaluated and here the trends were found to be similar independent from the class of substance. The lower the peak height, the larger was the observed SD, more or less independently from the investigated compound. For the n-alkanes, the increase was in parallel for all five compounds (Figure S2, upper part) and overall less distinctive than for the investigated steroids. But here the signal intensities did not reach the very low values investigated for steroids. Within the steroids (Figure S2, lower part), ANDRO seems to be less affected but with the data at hand no significantly different behavior could be verified. In the context of doping control analysis, the obtained results clearly demonstrate the possibility to investigate steroids at low urinary nanogram-per-milliliter concentrations resulting in low signal intensities as long as the suitability of the complete analytical procedure was carefully validated. And suitable instrument performance for each investigated sample has to be ensured by, for example, quality control samples at similar concentration to the investigated sample or standard steroid injections at different concentrations to prove the instrument linearity. These measures are good practice in doping control analysis and their necessity is only highlighted once more by the presented results.
Reference population-based values
Absolute δ13CVPDB-values
The CIR found within this population mainly reflect the expected values for a European population [4, 5, 7, 10, 11, 14]. The results are depicted in Fig. 2 illustrating that different steroids exhibit significantly different median values (t test, p < 0.05). Sorting of all steroids in ascending order by their CIR results in a distinctive pattern. Direct comparison with recently published data [10, 14] shows that different methods or different investigated populations result in non-conformities regarding the pattern of herein measured CIR. For example, TESTO was found more depleted than 5aDIOL in this study (Fig. 2) but de la Torre et al. observed 5aDIOL to be more depleted than TESTO [14]. In addition, ETIO and ANDRO commonly yield a Δ value around 1 ‰; however, Sobolevsky et al. found equal values for both steroids [10]. In these cases with the data at hand, it is impossible to elucidate if sample preparation or the constitution of the reference population is responsible for the measured differences.

Box plot of δ13CVPDB-values obtained for the investigated reference population (n = 67). ERC endogenous reference compounds (excreted as glucuronides), GLUC target analytes excreted as glucuronides; SULF target analytes found as sulfoconjugates in urine
However, a more detailed comparison seems possible for the values obtained in this study to the published ones using a comparable reference population and method [5, 11]. All available values for comparison have been brought face to face in Table 2. Obviously, a stable offset between the data of approximately 1.4 ‰ is visible. This might be due to the fact that secondary isotopic reference material in terms of steroids was not available in 2008.
Regarding the above-mentioned pattern, the main difference for the glucuronidated steroids can be found for both 5aDIOL and 5bDIOL together with EPIT. While the result for EPIT is not surprising and similar to the outcome described for a different reference population studied with focus on EPIT and its metabolites [9] the change in both 5a- and 5bDIOL was surprising. In contrast to the older method where 5aDIOL was more depleted than 5bDIOL now both steroids showed similar values. Most probably, this is due to the enhanced clean up as for the older method a weak correlation was found between the CIR of both steroids and the concentration ratio of 5bDIOL/5aDIOL emerging from incomplete separation of both steroids on the HPLC. Small portions of 5bDIOL could contribute to the fraction collected for 5aDIOL depleting the CIR of this steroid according to chromatographic isotopic fractionation on a reversed-phase column [30, 31]. This effect was circumvented by the modified HPLC conditions and differences in fraction collection as performed with the enhanced method. In accordance with the complete separation of both steroids no correlation between CIR and steroid concentrations was found anymore. This also affected the calculated Δ-values and thresholds as discussed later on.
Interestingly, on the other steroids only differing in their 5α- and 5β-configuration, ETIO and ANDRO, which were also not completely separated on neither the HPLC nor the GC column the new method did not have such an impact. Besides, the offset ANDRO shows slightly less-enriched values comparing to ETIO which might agree with the missing crosswise influence of both steroids eluting closely from the GC column.
Considering sulfoconjugated steroids, a similar trend for ETIO and ANDRO was visible. The most surprising value was found for EPIA which might be explained by co-eluting ETIO as was also detectable for HIR of this steroid [13].
Even if the factors influencing the measured CIR values cannot be attributed unambiguously, the results clearly demonstrate the possibility of method parameters influencing the obtained results in both δ- and Δ-values. As a consequence, altered sample preparation protocols necessitate careful assessment and if any impact is ascertainable, a repeated determination of reference-based thresholds seems inevitable. These considerations also support the idea of reference-based values in contrast to fixed thresholds as systematic offsets evoked by the used method cannot be precluded.
Δ-values and reference limits
In the context of doping control analysis, the differences between an ERC and an appropriate TC proved to be the most reliable and sensitive marker to detect misuse of steroids. As these will be the important ones for doping control analysis, all thresholds calculated for glucuronidated steroids are listed in Table 3. Besides, the common combinations of ERC and TC also the calculated values obtained with DHEA as ERC have been listed as in case of a TESTO administration or any use of a pro-hormone with an androst-4-ene structure DHEA will not be influenced and can therefore serve as ERC.
Besides the above-mentioned changes in mean Δ values due to difference in δ values, the thresholds established within this study are slightly improved as SDs were found lower especially for the lesser concentrated urinary steroids. This should enhance the sensitivity of the method which can further be improved by combination of CIR and HIR measurements as will be discussed later on.
Factors influencing CIR
Several factors influencing the CIR of endogenous steroids have been identified in previous studies [4-6, 32]. Within the present study, a significant difference (t test, p < 0.001) was detected only between steroids excreted glucuronidated or sulfoconjugated. Interestingly, this effect is seen on both CIR and HIR values and shows comparable values for all three investigated steroids (ETIO, ANDRO, and DHEA). The difference in CIR and HIR is ca. 1 ‰ and 11 ‰, respectively [13]. In both cases, the sulfoconjugated analyte shows the more enriched values. A possible explanation might be different isotopic values at different steroid production sites within the body [18].
Differences between the genders were not detected; this may be attributed to the fact that out of 29 investigated females, only seven used oral contraceptives, which are presumed to be responsible for gender-related differences [4, 32].
Excretion study
The established reference limits have been tested for by a single oral administration of TESTO-undecanoate. The liberated TESTO was found at δ13CVPDB = −27.04 ± 0.32 ‰ (n = 24) and the ester at δ13CVPDB = −28.46 ± 0.23 ‰ (n = 6).
All investigated TC are strongly depleted after administration. In accordance to literature data, not all metabolites of TESTO were found equally influenced [5, 33-36]. Nevertheless, the detection windows for ANDRO, ETIO, 5aDIOL, and TESTO itself where comparable between 12 and 18 h. Only 5bDIOL was influenced for a longer time period and found elevated above the established threshold for more than 30 h. These detection windows are at least comparable to the already published ones and prove the usefulness of the reference-based decision limits.
Comparing the results obtained for CIR to those acquired for HIR reveals an interesting point. While both methods seem to yield over all comparable results, TESTO was less influenced in HIR. The TESTO excreted into urine directly after application is assumed to derive more or less completely from the exogenous source. Therefore, the isotopic ratios should equal the administered steroid. This holds true for CIR values where the urinary TESTO with δ13CVPDB = −27.14 ± 0.20 ‰ (n = 5) perfectly reflects the value found for the liberated steroid as mentioned above. However, for HIR, a large discrepancy was detected with urinary TESTO at δ2HVSMOW = -255.0 ± 7.7 ‰ (n = 5) and the related liberated standard showed a value of δ2HVSMOW = -208.2 ± 4.7 ‰ (n = 24) [13]. As the CIR values prove that the endogenous contribution of TESTO is negligible a sound explanation for the significant difference (p < 0.001, t test) in HIR seems not possible. An isotopic fractionation during the saponification of the steroid ester, regardless if for the chemical cleavage in vitro or the enzymatic cleavage in vivo, only affecting HIR but not CIR, is conceivable as the steroid carbon backbone is not involved while hydrogen exchange might occur. A more reasonable explanation might be the rapid inter-conversion of TESTO with androst-4-ene-3,17-dione which might enable introduction of depleted hydrogen from NADPH [37, 38]. The data at hand does not allow to draw any conclusion and this topic should be investigated further on.
Combining CIR and HIR
The results obtained during the investigations on HIR revealed the possibility to use hydrogen isotopes complementary to carbon isotopes for doping control purposes. A sample with a high urinary TESTO concentration showed no suspicious CIR values in its TC but clearly in the HIR values [13]. Besides this independent use of both methods, it should be possible to benefit from the combination of both stable isotopes in sports drug testing. As demonstrated on both samples from the reference population and the excretion study it is easily possible to determine isotopic ratios for both elements using only one sample preparation. As the HIR measurements necessitate approx. 10 times more steroid material than CIR determinations, the remnant conserved after injecting the sample for HIR can be reconstituted and forwarded to CIR evaluation. Within the reference population investigations, only 21 out of 670 re-injections for CIR of glucuronidated steroids could not be completed due to insufficient amounts of analyte/sample and within the excretion study no measurement failed.
Reference population-based values
Both δ- and Δ-values did not show any significant (p < 0.1) correlation between HIR and CIR which is also visible in the example depicted in Fig. 3. Here the Δ-values of PD-ANDRO are plotted exemplarily to discuss the possible benefit of combining both stable isotopes. Regarding CIR, currently the WADA-defined threshold of 3 ‰ has to be applied to doping control samples. As this threshold does not fit for each pair of steroids, reference population derived limits are more beneficial. Usually the threefold standard deviation is added to the mean value to calculate the 99.7 % reference limit. The same applies for HIR values.

Δ-values of PD-ANDRO, CIR plotted against HIR. The ellipse represents the parametrically calculated 99.7 % reference interval and the solid lines the 95 % interval for each stable isotope. Further information in the text
As both isotope ratio determinations are independent from each other and as long as there is no covariance between the data, the two-dimensional distribution density will result in an ellipse with the parameters a and b defined by the SD from the one-dimensional distributions. So the resulting confidence area with 99.7 % as plotted in Fig. 3 will reflect the threefold standard deviation found for HIR when CIR are found at mean values and vice versa. If a Δ-value for CIR falls near the determined reference limit the deviation in HIR can be as small as a single SD to result in a finding not in accordance with endogenous steroid production.
An easy-to-handle approach might be to use of 95 % reference limits (by adding the twofold SD) for both isotopes simultaneously resulting in an approximate over all statistical certainty of also 99.75 %. The approach of using both stable isotopes should lead to either a higher statistical significance to separate endogenous from exogenous steroids, or when lowering the reference limits as suggested, to a prolonged detection time for steroid abuse. If one of both isotopes is within the reference interval, for the other the usual threshold adding the threefold SD should be applied.
Excretion study
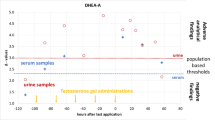
The results after oral administration of TESTO-undecanoate show a prolonged detection time if the above-mentioned rule is applied. Especially for PD-ETIO changing from 18 to 32 h the difference is clearly visible (Fig. 4). Directly after the administration, both HIR and CIR values are strongly influenced until 6 h after administration. Then the endogenous steroid production start to superimpose the exogenous influence on both Δ values and they start to decline back to normal values. By application of the combined 95 % reference limits, all five measurements around sample 15 are still clearly outside the limits and would constitute a doping violation. By focusing only on CIR or HIR, these values covering the post-administration period from 18.2 to 32.5 h would have to be declared negative.

Δ-values of PD-ETIO after administration of 100 mg TESTO orally, CIR plotted against HIR. The dashed lines represent the 95 % reference limits, the arrows illustrate the course of values post administration. 1 blank urine, 6 6.5 h after administration, 9 11.5 h after administration, 15 32.5 h after administration, 16 37.8 h after administration
For other steroids, the change is not as pronounced but still noticeable like for PD-ANDRO (from 11 to 18 h). Unfortunately, with only one excretion study at hand, it will be difficult to judge if a reasonable prolongation emerges or not. Further studies are recommended to estimate the prolongation of the detection window for TESTO misuse.
Metabolism studies
The combination of both stable isotopes might also be of interest in metabolism studies on endogenous steroids. As depicted in Fig. 5, every steroid shows a distinctive value on the two-dimensional CIR vs. HIR plot. Evaluating the natural, uninfluenced values raises some interesting questions. As already mentioned, all the sulfoconjugated steroids show more enriched values in both isotopes—with the exception of EPIA. Regarding its CIR, it is even more depleted than glucuronidated ANDRO and has a similar HIR value. The fact that this steroid might have a unique place in the metabolism was already discussed in the context of a DHEA administration study where it showed prolonged influence compared to all other steroids [11]. This might be due to a certain compartmentalization of this steroid or the involved 3β-hydroxylase. Another nonconformity within the sulfated steroids show both 5ENDIOLs with values far from DHEA-sulfate although they are closely related to each other in the metabolism by 17-hydroxysteroid-dehydrogenase activity and can be converted into each other.

Scatter plot of mean δ13CVPDB-values against mean δ2HVSMOW-values. The error bars depict the average singe standard deviation, steroids excreted sulfated into urine are marked with (_S)
Regarding HIR, especially the steroids with 5α- versus 5β-conformation show a noticeable pattern. The 5α-steroids ANDRO, ANDRO_S, and 5aDIOL are always around 15 ‰ more enriched than the 5β-steroid analogues ETIO, ETIO_S, and 5bDIOL. In parallel, the CIR differ by approx. 1 ‰ for ETIO and ANDRO. The DIOLs however show similar values here which might be assignable to the relatively large contribution of TESTO to 5aDIOL as both show nearly similar values.
These preliminary considerations about the isotopic two-dimensional distribution pattern of steroids are intended to highlight the capabilities of combining the analytical results of two different stable isotopes. This might further become an interesting point for excretion studies with two different compounds labeled with different stable isotopes and administered simultaneously.
Conclusion
The already existing sample preparation method to determine CIR of urinary steroids has been improved and according to the guidelines of the International Federation of Clinical Chemistry and Laboratory Medicine a new reference population was investigated in order to establish appropriate decision limits for sports drug testing. As expected, the reference limits for 5aDIOL and 5bDIOL changed slightly and demonstrated the necessity of new threshold determinations after method changes.
Within this study, the first simultaneously determination of HIR and CIR as well as glucuronidated and sulfated steroid enabled unprecedented comparison and revealed distinctive patterns within the examined distributions. In general, sulfated steroids are more enriched in both carbon and hydrogen isotopes for relatively stable values supporting the already hypothesized difference in isotopic ratios at different endogenous production sites. Nevertheless, the scatter plot of mean HIR versus mean CIR values illustrates that additional effects like isotopic fractionation or compartmentalization might occur during steroid metabolism. Ongoing research should benefit from the potentials two-dimensional isotopic investigations can offer.
The combination of both isotopes allows for lower reference limits providing similar statistic power to distinguish between endogenous and exogenous steroids. Additional excretion studies will be necessary to evaluate the benefit doping control might derive by this novel approach.
References
Ueki M, Okano M (1999) Analysis of exogenous dehydroepiandrosterone excretion in urine by gas chromatography/combustion/isotope ratio mass spectrometry. Rapid Commun Mass Spectrom 13:2237–2243
Aguilera R, Chapman TE, Starcevic B, Hatton CK, Catlin DH (2001) Performance characteristics of a carbon isotope ratio method for detecting doping with testosterone based on urine diols: controls and athletes with elevated testosterone/epitestosterone ratios. Clin Chem 47:292–300
Aguilera R, Hatton CK, Catlin DH (2002) Detection of epitestosterone doping by isotope ratio mass spectrometry. Clin Chem 48:629–636
Flenker U, Güntner U, Schänzer W (2008) δ13C-values of endogenous urinary steroids. Steroids 73:408–416
Piper T, Mareck U, Geyer H, Flenker U, Thevis M, Platen P, Schänzer W (2008) Determination of 13C/12C ratios of endogenous urinary steroids: method validation, reference population and application to doping control purposes. Rapid Commun Mass Spectrom 22:2161–2175
Cawley AT, Trout GJ, Kazlauskas R, Howe CJ, George AV (2009) Carbon isotope ratio (δ13C) values of urinary steroids for doping control in sport. Steroids 74:379–392
Piper T, Flenker U, Mareck U, Schänzer W (2009) 13C/12C ratios of endogenous urinary steroids investigated for doping control purposes. Drug Test Anal 1:65–72
Strahm E, Emery C, Saugy M, Dvorak J, Saudan C (2009) Detection of testosterone administration based on the carbon isotope ratio profiling of endogenous steroids: international reference populations of professional soccer players. Br J Sports Med 43:1041–1044
Piper T, Riemann P, Opfermann G, Mareck U, Geyer H, Vajiala G, Flenker U, Schänzer W (2009) Determination of 13C/12C ratios of urinary epitestosterone and its main metabolites 5α- and 5β-androstane-3α,17α-diol. Drug Test Anal 1:576–586
Sobolevsky T, Prasolov IS, Rodchenkov G (2010) Carbon isotope mass spectrometry in doping control. J Anal Chem 65:825–832
Piper T, Opfermann G, Thevis M, Schänzer W (2010) Determination of 13C/12C ratios of endogenous urinary steroids excreted as sulpho conjugates. Rapid Commun Mass Spectrom 24:3171–3181
Piper T, Fusshöller G, Emery C, Schänzer W, Saugy M (2012) Investigations on carbon isotope ratios and concentrations of urinary formestane. Drug Test Anal 4:942–950
Piper T, Thomas A, Thevis M, Saugy M (2012) Investigations on hydrogen isotope ratios of endogenous urinary steroids: reference population based thresholds and proof-of-concept. Drug Test Anal 4:717–727
de la Torre X, Colamonici C, Curcio D, Molaioni F, Botre F (2012) A comprehensive procedure based on gas chromatography-isotope ratio mass spectrometry following high performance liquid chromatography purification for the analysis of underivatized testosterone and its analogues in human urine. Anal Chim Acta 756:23–29
Coplen TB (2011) Guidelines and recommended terms of expression of stable-isotope-ratio and gas-ratio measurement results. Rapid Commun Mass Spectrom 25:2538–2560
World Anti-Doping Agency: TD2004EAAS (2013) http://www.wada-ama.org/Documents/World_Anti-Doping_Program/WADP-IS Laboratories/Technical_Documents/WADA_TD2004EAAS_Reporting_Evaluation_Testosterone_Epitestosterone_TE_Ratio_EN.pdf. Accessed 02 Feb 2013
Michal G (1999) Biochemical pathways. Spektrum Akademischer, Heidelberg
Piper T, Baume N, Strahm E, Emery C, Saugy M (2012) Influences of β-HCG administration on carbon isotope ratios of endogenous urinary steroids. Steroids 77:644–654
Solberg HE (1993) A guide to IFCC recommendations on reference values. J Int Fed Clin Chem 5:162–166
Solberg HE. The IFCC recommendation on estimation of reference intervals. The RefVal Program. Clin Chem Lab Med 42:710–714 (and references therein)
Zhang Y, Tobias HJ, Brenna JT (2009) Steroid isotopic standards for gas chromatography-combustion isotope ratio mass spectrometry (GCC-IRMS). Steroids 74:369–378
Cawley A, Collins M, Kazlauskas R, Handelsman DJ, Heywood R, Longworth M, Arenas-Queralt A (2010) Stable isotope ratio profiling of testosterone preparations. Drug Test Anal 2:557–567
Werner RA, Brand WA (2001) Referencing strategies and techniques in stable isotope ratio analysis. Rapid Commun Mass Spectrom 15:501–519
Docherty G, Jones V, Evershed P (2001) Practical and theoretical considerations in the gas chromatography/combustion/isotope ratio mass spectrometry δ13C analysis of small polyfunctional compounds. Rapid Commun Mass Spectrom 15:730–738
Forsdahl G, Östreicher C, Koller M, Gmeiner G (2011) Carbon isotope ratio determination and investigation of seized testosterone preparations. Drug Test Anal 3:814–819
Kaunzinger A, Juchelka D, Mosandl A (1997) Progress in the authenticity assessment of vanilla. 1. Initiation of authenticity profiles. J Agric Food Chem 45:1752–1757
Cawley A, Flenker U (2008) The application of carbon isotope ratio mass spectrometry to doping control. J Mass Spectrom 43:854–864
Piper T, Geyer H, Gougoulidis V, Flenker U, Schänzer W (2010) Determination of 13C/12C ratios of urinary excreted boldenone and its main metabolite 5β-androst-1-en-17β-ol-3-one. Drug Test Anal 2:217–224
de la Torre X, Colamonici C, Curcio D, Molaioni F, Pizzardi M, Botre F (2011) A simplified procedure for GC/C/IRMS analysis of underivatized 19-norandrosterone in urine following HPLC purification. Steroids 76:471–477
Braunsdorf R, Hener U, Przibilla G, Piecha S, Mosandl A (1993) Analytische und technologische Einflüsse auf das 13C/12C-Isotopenverhältnis von Orangenöl-Komponenten. Z Lebensm Unters Forsch 197:24–28
Filer CN (1999) Isotopic fractionation of organic compounds in chromatography. J Labelled Cdp Radiopharm 42:169–197
Piper T, Emery C, Saugy M (2011) Recent developments in the use of isotope ratio mass spectrometry in sports drug testing. Anal Bioanal Chem 401:433–447
Korenman SG, Wilson H (1966) On the role of androstendione and testosterone as precursors of urinary androsterone and 5β-androsterone. Steroids 8:729–737
Robel P, Emiliozzi R, Baulieu EE (1966) Studies on testosterone metabolism. III. The selective “5β-metabolism” of testosterone glucuronide. J Biol Chem 241:20–29
Mauvis-Jarvis P, Floch H, Jung I, Robel P, Baulieu EE (1968) Studies on testosterone metabolism. VI. Precursors of urinary androstanediols. Steroids 11:207–224
Peng SH, Segura J, Farré M, Gonzalez JC, de la Torre X (2002) Plasma and urinary markers of oral testosterone undecanoate misuse. Steroids 67:39–50
Horton R, Tait JF (1966) Androstenedione production and interconversion rates measured in peripheral blood and studies on the possible site of its conversion to testosterone. J Clin Investig 45:301–313
Schmidt HL, Werner RA, Eisenreich W (2003) Sytematics of 2H patterns in natural compounds and its importance for the elucidation of biosynthetic pathways. Phytochem Rev 2:61–85
Acknowledgments
This project was partly funded by the Federal Ministry of the Interior of the Federal Republic of Germany and the Manfred Donike Institute for Doping Analysis, Cologne.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 165 kb)
Rights and permissions
About this article
Cite this article
Piper, T., Emery, C., Thomas, A. et al. Combination of carbon isotope ratio with hydrogen isotope ratio determinations in sports drug testing. Anal Bioanal Chem 405, 5455–5466 (2013). https://doi.org/10.1007/s00216-013-6949-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-6949-3