
Abstract
An automated flow-through multi-mycotoxin immunoassay using the stand-alone Munich Chip Reader 3 platform and reusable biochips was developed and evaluated. This technology combines a unique microarray, prepared by covalent immobilization of target analytes or derivatives on diamino-poly(ethylene glycol) functionalized glass slides, with a dedicated chemiluminescence readout by a CCD camera. In a first stage, we aimed for the parallel detection of aflatoxins, ochratoxin A, deoxynivalenol, and fumonisins in cereal samples in a competitive indirect immunoassay format. The method combines sample extraction with methanol/water (80:20, v/v), extract filtration and dilution, and immunodetection using horseradish peroxidase-labeled anti-mouse IgG antibodies. The total analysis time, including extraction, extract dilution, measurement, and surface regeneration, was 19 min. The prepared microarray chip was reusable for at least 50 times. Oat extract revealed itself as a representative sample matrix for preparation of mycotoxin standards and determination of different types of cereals such as oat, wheat, rye, and maize polenta at relevant concentrations according to the European Commission regulation. The recovery rates of fortified samples in different matrices, with 55–80 and 58–79 %, were lower for the better water-soluble fumonisin B1 and deoxynivalenol and with 127–132 and 82–120 % higher for the more unpolar aflatoxins and ochratoxin A, respectively. Finally, the results of wheat samples which were naturally contaminated with deoxynivalenol were critically compared in an interlaboratory comparison with data obtained from microtiter plate ELISA, aokinmycontrol® method, and liquid chromatography–mass spectrometry and found to be in good agreement.

Principle of the competitive chemiluminescence ELISA using the microarray chip
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The mycotoxins are secondary metabolites from fungal species such as Aspergillus, Fusarium, Alternaria, and Penicillium growing on agricultural commodities in the field or during storage [1]. Aflatoxins mainly occur in rice, nuts, cotton seed, corn, and other cereals, while ochratoxin A is mainly found in coffee, rice, wine, beer, spices, and particularly also in crops and cereals [2, 3]. Trichothecenes such as deoxynivalenol (type B) or T-2 toxin (type A) are produced in cereals, respectively, but contamination of fruits and vegetables has also been reported [4]. Furthermore, fumonisins are mainly found in corn and corn-based products [4]. These mycotoxins are known for their acute toxic, immunosuppressive, mutagenic, teratogenic, or even carcinogenic effects. They are of low molecular weight and rather stable, which makes it nearly impossible to remove them from foodstuffs for example by thermal processing or variation of pH [5, 6]. Since they represent a potential health hazard to humans and animals, maximum levels for several mycotoxins in food have been set by the European Community (EC) [7, 8]. For example, maximum levels of 4 μg kg−1 for the sum of aflatoxins B1, B2, G1, and G2 as well as 2 μg kg−1 for aflatoxin B1 and 5 μg kg−1 for ochratoxin A in cereals and all products derived from cereals are allowed by the commission regulations (EC) No 1881/2006 and No 1126/2007. In contrast, higher limits for fumonisin B1 of 400–4,000 μg kg−1 and for deoxynivalenol of 750–1,750 μg kg−1 were set. The reason is that aflatoxins and ochratoxin A are classified as carcinogenic, especially aflatoxin B1 is listed as a group 1 carcinogen by the International Agency for Research on Cancer [9].
Mycotoxins constitute a very heterogeneous group of compounds which differ greatly in their chemico-physical properties and polarity as well as their distribution and concentration in food (Fig. 1). Because of this, particular extraction, clean-up, and detection strategies have been developed. For the qualitative and quantitative analysis of mycotoxins in food traditionally high-performance liquid chromatography (HPLC), gas chromatography (GC) and thin-layer chromatography (TLC) are used ([10–12]; and references therein). TLC is mostly used for a rapid sample screening and is considered as sufficiently selective for single target analysis. GC methods in combination with ECD, FID, or mass spectrometry detection are mainly described for the determination of trichothecenes. The most common method for routine analysis of mycotoxins in food is HPLC. HPLC detection using DAD or FLD, due to the presence of a chromophore, is possible directly (e.g., ZEA, OTA) or requires pre- or post-column derivatisation (e.g., aflatoxins and fumonisins). Over the last decade, HPLC hyphenated to tandem mass spectrometry (LC-MS/MS) has become the method of choice owing to the development of efficient electrospray (ESI) and atmospheric pressure chemical ionization interfaces [13–16]. However, an entire sample preparation and especially extract clean-up is still required using, e.g., solid phase extraction (SPE) based on conventional reversed-phase materials, mixed-mode cartridges (MycoSep®), molecularly imprinted polymers, and immunoaffinity columns [17–18]. Even so, reliable quantification can often be achieved only by matrix-matched calibration or use of isotope-labeled internal standards [11, 17, 19], although the “dilute-and-shoot” multianalyte approaches without any sample cleanup after extraction represent a rapid method which might be suitable for several matrices analyzed by LC-MS/MS [20].

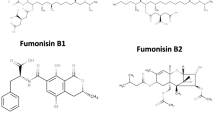
The chemical structures of aflatoxins B1, B2, G1, G2, deoxynivalenol, fumonisin B1, and ochratoxin A
To provide rapid and sensitive detection based on cost-effective and easy to use methods, which can be performed in a non-laboratory environment by non-specialists, a multitude of the so-called screening methods was developed. Generally, they do not require sample preparation other than extraction and dilution; however, they are less precise and sensitive than chromatographic methods. This field is dominated by immunochemical tests which use anti-mycotoxin antibodies in a variety of different assay formats like, e.g., enzyme-linked immunosorbent assay (ELISA), fluorescence polarisation immunoassay (FPIA), and kinetic FPIA, also called stopped-flow FPIA (aokinmycontrol® kits, Aokin AG, Berlin, Germany) [6, 10, 12, 21, 22]. Notably, the need to provide even more simple, fast, robust, user-friendly, and cost-effective methods has led to a multitude of non-instrumental membrane-based lateral flow devices, often incorrectly referred to as “dipsticks,” and gel-based flow-through tests with steadily improved detection capability and use of different labels [23–27]. Normally, these tests provide a “yes/no” result for a certain limit or range set previously; however, during the last years, there is a tendency to develop modified devices enabling quantitative evaluation and multiplexing ([28–31]; and references therein). In addition, a few emerging non-immunochemical methods exist which need only sieving and grinding of the samples. They exploit several spectroscopic techniques like Fourier transform mid-infrared spectroscopy attenuated total reflection or two-photon-induced fluorescence [32, 33].
Because multimycotoxin methods are highly desirable in order to keep analysis time and costs low, the biosensor development increasingly focuses on parallel analysis of several mycotoxins [34–36]. One of the most promising techniques is the MultiAnalyte Profiling (xMAP) technology from Luminex (Austin, TX) which combines a unique color-coded microsphere suspension array with a dedicated flow cytometer. Using this technique, six mycotoxins were detected in parallel [37]. Another useful label-free analytical technique might be imaging surface plasmon resonance-based multiplex microimmunoassay platform from IBIS Technologies (Enschede, The Netherlands). The sensor chip has perspective for the determination for up to 40 different mycotoxins [38]. In this work, an indirect competitive immunoassay on regenerable, reusable glass microchips for the parallel determination of four mycotoxins, i.e., aflatoxins, ochratoxin A, deoxynivalenol, and fumonisin B1 in cereal extracts on a fully automated flow-through device with chemiluminescence readout was developed. The automated system (MCR 3) was previously applied, beside others, for the parallel detection of 13 antibiotics in milk and ochratoxin A in green coffee extracts [39–41].
Experimental
Chemicals and materials
All standard chemicals and reagents were obtained from Sigma-Aldrich (Steinheim, Germany). The chemiluminescence substrate solutions (SuperSignal ELISA Femto Maximum Sensitivity Kit) were obtained from Pierce (Rockford, USA). Diamino-poly(ethylene glycol) (DAPEG) was a gift from Huntsman Holland (Rozenburg, The Netherlands). 3-Glycidyloxypropyltrimethoxy-silane, 1-hydroxybenzotriazole hydrate (HOBt), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N,N-diisopropylethylamine (DIPEA), and sodium dodecyl sulfate (SDS) were obtained from Sigma-Aldrich. Ochratoxin A (OTA) standard (10 μg mL−1) and deoxynivalenol (DON) standard (100 μg mL−1) both in acetonitrile were purchased from LGC Standards (Wesel, Germany) and used for the calibrations. Solid OTA, fumonisin B1 (FB1), and aflatoxins B1 (AFB1), B2, G1, and G2 were obtained from Sigma-Aldrich. Aflatoxin standards in acetonitrile were prepared as described elsewhere [19]. Mouse monoclonal anti-OTA antibody 5G9 was acquired from Soft Flow Biotechnology (Gödöllö, Hungary). The generation and characterization of mouse monoclonal anti-aflatoxin antibody 1F2 was described elsewhere [42]. Horseradish peroxidase-labeled anti-mouse IgG produced in horse was purchased from Axxora (Loerrach, Germany). Buffers and solutions were prepared freshly in ultrapure water which was obtained by reverse osmosis with UV treatment (Milli-RO 5 Plus, Milli-Q185 Plus, Millipore, Eschborn, Germany). Conventional microscopy glass slides (26 × 76 × 1 mm) were obtained from Carl Roth (Karlsruhe, Germany). The carriers of the microarray flow cells were made from black poly(methyl methacrylate) and were produced in-house. The adhesive foil ARcare 90106 for the connection of glass slides and plastic carriers was supplied by Adhesive Research Ireland Ltd. (Limerick, Ireland). The laser cut of the adhesive foil was carried out by A.L.L. Lasertechnik GmbH (München, Germany). Different kinds of flours (oat, wheat, rye) and maize polenta were acquired in supermarkets in München or made available from Rosenmühle (Landshut, Germany).
Indirect microplate-based ELISA
The microtiter plates (655061, Greiner, Frickenhausen, Germany) were washed automatically with a 96-channel plate washer (ELx405 Select), and the absorbance was measured with a microtiter plate reader (Synergy HT) both from Bio-Tek (Bad Friedrichshall, Germany). For the indirect microplate-based ELISA, 200 μL per well of a 1:10,000 dilution in coating buffer of an OTA-BSA (1 mg mL−1) and FB1-BSA conjugate (1 mg mL−1), and a 1:5,000 dilution in coating buffer of AFB1-BSA (1 mg mL−1) and DON-HS-BSA conjugate (1 mg mL−1) were incubated overnight at 4 °C. The coating buffer (pH 9.6) consisted of 1.6 g disodium carbonate, 2.9 g sodium hydrogen carbonate, and 0.2 g sodium azide in 1 L of water. After a threefold washing step with washing buffer, the plates were incubated with 1 % casein (300 μL/well, shaken at 300 rpm) in PBS (w/v) for 1 h at room temperature. For the PBS, 1.4 g potassium dihydrogen phosphate, 12.2 g dipotassium hydrogen phosphate, and 8.5 g sodium chloride in 1 L of water were used. The washing buffer consisted of 42 mL washing buffer concentrate (8.2 g potassium dihydrogen phosphate, 73.2 g dipotassium hydrogen phosphate, 52.6 g sodium chloride, and 30 mL Tween 20 in 1 L of water) in 2.5 L of water. For the competition reaction, the toxin calibration solution (100 μL/well) and the primary antibody diluted in PBS (100 μL/well) were added, and the plates were shaken at 100 rpm for 1 h at room temperature. A 1:5,000 dilution of the anti-OTA (1 mg mL−1) and anti-DON (0.72 mg mL−1) antibodies and a 1:10,000 dilution of the anti-aflatoxin (1 mg mL−1) and anti-FB1 (0.37 mg mL−1) antibodies in PBS were used. For the construction of calibration curves, AFB1 and OTA at concentrations of 0.005, 0.01, 0.05, 0.1, 0.5, 1, 5, 10 μg L−1, FB1 at concentrations of 0.2, 0.5, 2, 5, 20, 50, 200, 500 μg L−1, and DON at concentrations of 0.1, 0.5, 1, 5, 10, 50, 100, 500 μg L−1 in oat extract were used. After another washing step, the plate was incubated with HRP-labeled anti-mouse antibody (dilution 1:8,000 in PBS, 200 μL/well) at 100 rpm for 1 h at room temperature. Next to the last washing step, substrate solution (200 μL/well) was added and incubated at 100 rpm at room temperature. The substrate buffer (pH 3.8) was prepared with 46.0 g potassium dihydrogen citrate and 0.1 g potassium sorbate in 1 L of water. The substrate solution consisted of 25 mL substrate buffer, 500 μL TMB stock solution (375 mg of 3,3′,5,5′-tetramethylbenzidine in 30 mL of DMSO), and 200 μL 1 % hydrogen peroxide. The color development was controlled visually and was stopped after 10 min by adding 5 % H2SO4 (100 μL/well). Finally, the absorbance was measured with the plate reader at 450 nm.
Synthesis of aflatoxin B2-carboxymethyl oxime and DON-hemisuccinyl
AFB2 was immobilized on the chip surface via a carboxyl group after functionalization with a carboxymethyl oxime spacer as described elsewhere [43]. The product was purified by semi-preparative HPLC and characterized by LC-ESI-MS (m/z = 388.10 [M + H]+). The DON was functionalized with succinyl anhydride as published before [44].
Fabrication of the glass slides
The DAPEG-functionalized glass slides were fabricated as described [45]. To remove all contaminants from the glass surface, the slides were incubated with 2 % Hellmanex (Hellma GmbH & Co. KG, Müllheim, Germany) under shaking overnight at room temperature. Furthermore, they were cleaned by sonication with Hellmanex and washed intensively with Millipore water. In the next step, the glass slides were treated with a freshly prepared MeOH/37 % HCl (1:1, v/v) for 1 h under shaking. After washing with Millipore water, the slides were shaken for another 1 h with concentrated sulphuric acid. Afterwards, they were cleaned with water and dried under N2 flow. The pre-treated glasses were silanized with GOPTS. Therefore, 600 μL GOPTS was added on the chip surface and covered with a second slide (sandwich technique). The incubation time at room temperature took 3 h. The slides were separated from each other in pure ethanol and were cleaned by sonication subsequently with ethanol, methanol, and ethanol for 15 min. After drying under N2 flow, the slides were covered with 1 mL molten DAPEG and incubated at 100 °C for 15 h by using the sandwich technique again. At the end, the modified slides were separated and cleaned intensively with water by using sonication for two times each 15 min and then dried under N2 flow. They were stored for further use under vacuum and used within 1 month.
Microarray construction
The DAPEG-functionalized glass slides were directly applied to the spotting process, using a BioOdyssey Calligrapher Miniarrayer (Bio-Rad Laboratories GmbH, München, Germany) and the steel needle SNS 12 (Arrayit, Sunnyvale, USA). Aflatoxin B2-carboxymethyl oxime (AFB2-CMO), 2.5 μL, aqueous solution (10 mg mL−1) was dissolved in 45 μL of spotting buffer. The spotting buffer consisted of 300 μL DMSO, 690 μL carbonate buffer (0.290 g NaHCO3 and 0.159 g Na2CO3 in 100 mL water, pH 8.5), and 10 μL glycerol. Five microliters of OTA in acetonitrile (5.5 mg mL−1) and FB1 in methanol (10 mg mL−1), respectively, were dissolved each in 45 μL of spotting buffer. Twenty microliters of DON-hemisuccinyl (DON-HS; 10 mg mL−1) was mixed with 30 μL spotting buffer. As a negative control spotting buffer was used. To introduce an activated carboxyl group into AFB2-CMO, OTA, and DON-HS, 10 eq. EDC, 10 eq. HOBt, and 50 μL DIPEA were added to the spotting buffer containing the mycotoxin derivatives and incubated for 3 h under shaking at room temperature. Twenty-five microliters of each of the spotting solutions was added into different cavities of a 384-well low binding microtiter plate (no. 788161, Greiner, Frickenhausen, Germany) and aliquots spotted on the DAPEG glass slides in six replicates. During the spotting process, the chips were tempered to 20 °C, and the spotting chamber humidity was set to 50 %. The chips were incubated for 15 h at room temperature and then washed firstly with PBS and further cleaned by sonication with water for 15 min. Afterwards, the chips were dried under N2 flow. The microarray chips were stored under vacuum and used within 4 weeks.
Sample preparation
For the extraction, 25 g of flour was mixed with 5 g of sodium chloride and 100 mL of methanol/water (80:20, v/v) with the T25 Ultra-Turrax® disperser (IKA Labortechnik, Staufen, Germany) for 5 min while cooling with ice. Afterwards, the extract was filtered through a folded filter (210 mm, Macherey-Nagel, Düren, Germany), which was obtained from Carl Roth. The extracts were stored at −20 °C and analyzed within 24 h. For the measurements on the Munich Chip Reader (MCR 3), the extracts were diluted fourfold with PBS to give a final methanol content of 20 %, resulting to a 16-fold dilution of the mycotoxin concentration in the sample.
The samples which were used for the determination of the recovery rates were spiked with aflatoxins, OTA, FB1, and DON in 1 mL of methanol first, then mixed together and finally air-dried overnight in a light-protected fume hood. The spiked samples were then extracted in the same way as described above. The amount of the flour was reduced to 5 g per sample.
Performance of the automated chemiluminescence immunoassay
As a blocking and running buffer 0.5 % (w/v) casein in PBS was used. Before starting the measurements, 3 × 1 mL casein/PBS was pumped with 200 μL s−1 through the flow cell. This step is important to displace possibly existing air bubbles in the flow cell of the chip. The program for the automated chemiluminescence immunoassay on the chip is summarized as follows: In the first step, 1 mL of the running buffer is submitted to the chip with a flow speed of 500 μL s−1. Afterwards, 500 μL of sample and same volume of the mixture of primary antibodies solutions in running buffer were pumped simultaneously into an incubation loop at a flow rate of 60 μL s−1. The primary antibodies mixture consisted of volumes of 1:5,000 dilutions of the anti-OTA and volumes of 1:15,000 dilutions of anti-aflatoxin antibody solutions (both at 1 mg mL−1) as well as a 1:4,000 dilution of anti-FB1 (0.37 mg mL−1) and 1:3,000 dilution of anti-DON (0.72 mg mL−1) in running buffer. The sample/antibodies mixture was pushed over the chip with 1 mL of running buffer at a flow rate of 100 μL s−1. After a washing step with 2 mL of running buffer at a flow rate of 500 μL s−1, the chip was incubated with 1 mL of HRP-labeled secondary antibody (200 μL at a flow rate of 100 μL s−1 and followed by 800 μL at a flow rate of 10 μL s−1). The chip was washed again with 2 mL of running buffer at a flow rate of 500 μL s−1, and then, 400 μL of enzyme substrate was flown over the chip at a flow rate of 150 μL s−1. Afterwards, the flow was stopped, and an image was taken for 60 s by the CCD camera. After the recording, the chip was exhaustively regenerated using a total volume of 22.5 mL of regeneration buffer, consisting of 7.51 g glycine, 5.85 g sodium chloride, and 0.1 % (w/v) SDS in 1 L of water. The pH was adjusted to 3.0 with concentrated hydrochloric acid. A flow rate of 100 μL s−1 was applied. The overall assay time was 11 min.
Data evaluation
The text files generated by the CCD camera were automatically evaluated with the Spot Image Processor 0.4 (SIP 0.4, Karsunke Softwarebüro, Wolnzach, Germany). The software not only automatically eliminates electronic artifacts of the CCD camera but also identifies all spots regard signal intensity and consistency with the spotted grid. Those spots that do not fit one of these criteria are identified as outliers, and the integrated area is moved to the identified grid. This kind of outlier correction is possible because it is known that the uncertainty of the used spotting system is not more than 5 μm in the x–y plane. After the grid identification, SIP 0.4 integrates the pixel intensities within a square of a given side length around each spot centre. A 9 × 9 pixel square around each spot center, which is up to an area of 0.13 mm2, was integrated, and the average value over all pixels in this area is given as the spot intensity.
Results and discussion
Cross-reactivity
Highly important for the multiplex microarray is the characterization of the antibodies, especially in regard to their cross-reactivity (CR) to the used mycotoxins. Therefore, the CR for aflatoxins, OTA, FB1, and DON was investigated using the indirect ELISA format on microtiter plates. First, in single experiments the IC50 value for each aflatoxin (B1, B2, G1, G2) was determined using standards prepared in oat extract. The CR values were calculated by the quotient of the IC50 values, setting the CR of AFB1 to 100 %. The results demonstrate that the anti-aflatoxin antibody has a high CR with all aflatoxins and thereby can be used to determine the sum value. Because the CR of the anti-aflatoxin antibody was nearly the same for AFB1 (100 %) and AFB2 (103 %), the latter was chosen to be immobilized on the chip surface due to its lower toxicity. No binding of the anti-aflatoxin antibody with OTA, FB1, and DON was observed. Moreover, also the other antibodies used turned out to be specific for their corresponding mycotoxin, i.e., no CR was detected.
Microarray fabrication
The principle of the multiplex flow-through assay is based on the indirect competitive ELISA as shown in Fig. 2. The mycotoxins are covalently immobilized on the DAPEG-functionalized chip surface that was used without further activation in comparison to the previously described procedure [41]. In the first experiments, AFB2, DON, FB1, and OTA were spotted on the chip surface directly. While AFB2 and DON did not give a signal significantly higher than the negative control (data not shown), OTA and FB1 spots could be clearly detected owing to the available carboxyl groups in their molecules, which enabled the linkage to the DAPEG surface, obviously. To test whether the signal intensity could be enhanced further by activation of the carboxyl groups, OTA and FB1 were incubated for 3 h in the active-spotting buffer containing EDC and HOBt, before spotting. As a result, the signal of the activated OTA was nearly two times higher compared to the non-activated one. On the contrary, activated FB1 gave non-uniform spots which was obviously caused by intra-/inter-polymerisation owing to the linkage of several FB1 molecules through the available carboxyl and amino functions. Because of this finding, FB1 was spotted with normal spotting buffer, i.e., without activating reagents. AFB2 was derivatized by a method published elsewhere to obtain a carboxymethyl oxime (AFB2-CMO) [43]. Instead of DON, the DON-HS was used. Both derivatives were then also coupled to the chip surface after activation of the carboxylic acid functions. Finally, acceptable signal intensities were obtained.

Principle of the indirect competitive chemiluminescence ELISA using the microarray chip. The mycotoxins are immobilized in a defined grid pattern. After primary and secondary antibodies bound to the chip surface, chemiluminescence reagents (luminol/H2O2) are added, and the light signal is detected with a CCD camera. The light intensity is inversely proportional to the concentration of mycotoxins in the sample
Regenerability
Because the chip production is a time-consuming procedure, it would be a great economic advantage if they could be used multiple times. This can only be achieved, if (a) all bound highly affine antibodies are completely removed, (b) biofouling of the chip surface by sample matrix components can be avoided, and (c) the spotted mycotoxins are chemically stable. In the past, a glycine regeneration buffer of pH value 3.0 and addition of an ionic surfactant, SDS (0.1 %), proved to be optimal and therefore was used in this study.
Measurements which were performed with only PBS as sample matrix after regeneration showed an almost constant maximal chemiluminescence signal for up to 50 measurements (data not shown). The same was observed for oat extract, respectively (Fig. 3). It can also be seen that especially the signals of the first measurements are always considerably lower compared to the following ones. It is supposed that the chip needs some conditioning to work properly, and therefore, three start-up cycles were run before calibration of the system. From the 4th to the 50th regeneration cycle, no significant signal loss could be observed. In detail, 96, 94, 92, and 88 % of the original signal intensity was obtained for OTA, AFB2, FB1, and DON-HS, respectively. After the 50th cycle, the relative standard deviation for the measurements was less than 6 %, confirming the stability and reproducibility of the chip surface and the assay. As the negative control (background), the spotting buffer was spotted to the chip surface. The investigation of the reusability provided two main general results. First, a time-consuming regeneration program with use of high volume of reagent (22.5 mL of regeneration buffer) was needed. Shorter regeneration programs with use of lower reagent volumes were not successful with real sample extracts. Second, the lower the density of immobilized mycotoxins on the chip surface, the easier is the regeneration. A reason for this observation might be that at higher densities, antibodies could bind to two immobilized antigen molecules on the chip surface and therefore were more difficult to remove. This problem is also known from surface plasmon resonance sensors with immobilized hapten molecules [46]. On the other hand, the higher the density of the immobilized mycotoxins, the higher are the obtainable chemiluminescence signals. Concluding, an optimized immobilization density has to be ascertained in order to balance reusability and adequate signal intensity.

The performance of 50 regeneration cycles using oat extract as a sample: ( ) aflatoxin B2, (
) aflatoxin B2, ( ) ochratoxin A, (
) ochratoxin A, ( ) Deoxynivalenol, (
) Deoxynivalenol, ( ) fumonisin B1, (
) fumonisin B1, ( ) negative control. The between-spot standard deviations (1 s; n = 6) are indicated as error bars
) negative control. The between-spot standard deviations (1 s; n = 6) are indicated as error bars
Dose–response curves
After the reusability of the multi-mycotoxin microarray was demonstrated, the dose–response curves of the flow-through indirect competitive ELISA were measured. For the preparation of standards, oat extract was fortified as described with four mycotoxins AFB1, OTA, FB1, and DON, respectively. The oat extract as sample matrix was chosen because this product constitutes baby food, which is subjected to rigorous inspection regarding possible contaminants, i.e., mycotoxins might be present, if any, at very low concentrations. The limit of detection (LOD) refers to the calculated analyte concentration corresponding to signal response of the blank plus three times of its standard deviation (SD). As working range, the linear range between 80 and 20 % of the maximum signal intensity was defined. The linear range for AFB1 was 0.16–0.73 μg L−1. Comprising a dilution factor of 16, this is equivalent to 2.6–11.6 μg kg−1. The LOD was 0.06 μg L−1 (0.9 μg kg−1). For OTA, the linear range of 0.11–0.96 μg L−1 (1.7–15.4 μg kg−1) and the LOD of 0.07 μg L−1 (1.1 μg kg−1) were calculated. For FB1, the linear range of 10.5–138.5 μg L−1 (168.1–2,215.8 μg kg−1) and LOD of 9.9 μg L−1 (159.0 μg kg−1) were obtained. Also, a much higher working range of 4.8–73.3 μg L−1 (76.2–1,173.1 μg kg−1) and LOD of 2.5 μg L−1 (40.5 μg kg−1) were ascertained for DON. It can be concluded that the obtained working ranges allow the measurement of all four relevant mycotoxins according to EC regulation.
Interday study
To test the intermediate precision of the new method, an interday study was carried out. Within 5 days, the dose–response curves for AFB1, OTA, FB1, and DON were carried out on three different chips of the same batch. The results are shown in Fig. 4 (Note: The chemiluminescence signals are not normalized, showing the high reproducibility of the assay). Although there is a tendency that the obtained maximum signal intensities were lowest with the chip measured on day 1, the performance of the assay was acceptable as is shown in Table 1. In this table, the IC50 values and linear ranges of the dose–response curves, indicated as amount of mycotoxin per kilogram of food sample, are summarized. The relative standard deviations (RSD) of the IC50 values of all mycotoxins were less than 10 %. The same applies to the lower and upper limits of the working ranges of OTA and DON standard curves. A slightly higher RSD of 12.5 % was found for the lower limit of the AFB1 linear range. For FB1, both the lower and upper limits with 15.4 and 18.5 % were highest. With another batch of microarray chips rather similar LODs and linear ranges were observed; however, the maximum signals were different. This indicates the fact that an optimized standardized chip production is urgently required to obtain high-quality chips. To this end, cleanrooms with controlled environment are indispensable.

Interday study for aflatoxin B1 (a), ochratoxin A (b), fumonisin B1 (c), and deoxynivalenol (d) carried out within 5 days. The between-spot standard deviations (1 s; n = 6) are indicated as error bars. Calibration curves were generated in oat extract
Recovery rates of fortified samples
To demonstrate the feasibility of the automated assay, dose–response curves prepared with oat extract were used to determine the recovery rates (RR) of fortified samples in different matrices, i.e., oat, wheat, and rye flour. Additionally, FB1 and DON were also measured in maize polenta samples. The results of the recovery experiments are shown in Table 2. All non-fortified samples could be confirmed as blanks, i.e., the determined concentrations were out of the working range or rather less than the LOD values. For the determination of RR, 5 g of oat, wheat, maize polenta, or rye flour was spiked with each of 1 μg kg−1of AFB1, 3 μg kg−1 OTA, 800 μg kg−1 FB1, and 400 μg kg−1 DON, to prepare samples which are contaminated with the mycotoxins at the MRL values or below as set by the EC. Additionally, another series of samples was spiked with higher concentration, i.e., each of 6 μg kg−1 AFB1, 7 μg kg−1 OTA, 1,000 μg kg−1 FB1, and 750 μg kg−1 DON. The calculated RR were 75–81 % for AFB1, 82–120 % for OTA, 55–80 % for FB1, and 58–79 % for DON, respectively. Differing RR of 140–152 % were obtained for the samples spiked with 1 μg kg−1 AFB1 which can be easily explained by the fact that this concentration is around the LOD value and clearly below the working range of the dose–response curve. Despite to this high calculated AFB1 concentration, the samples were determined correctly as negative samples, i.e., with a contamination level below the MRL. To investigate whether the determination of the sum value of aflatoxins is also possible in real samples, three different samples (oat, wheat, and rye) were spiked with 1.5 μg kg−1 of each of the four aflatoxins. The RR were between 127 and 132 % and therefore demonstrate the principal applicability of the microarray for the determination of the sum of relevant aflatoxins. One explanation for the lower recovery of DON and FB1 can be found in their reduced solubility in the extraction solvent used [methanol/water (80:20, v/v)]. Although the RR could be increased for these more hydrophilic mycotoxins by increasing the water content of the extractant (data not shown), a less polar solvent mixture has to be used for the extraction of all four relevant compounds in grain at an acceptable level.
Recovery rates of certified reference materials and naturally contaminated samples
The availability of certified reference materials is limited generally. For example, reference materials for aflatoxins in oat, wheat, or rye flour could not be obtained. For OTA, two wheat flour samples were purchased from IRRM, EU Joint Research Centre, Geel, Belgium (BCR®-471 with certified value < 0.6 μg kg−1) and Biopure, Tulln, Austria (2.7 ± 1.0 μg kg−1). Both reference materials were measured twice on two different days. The blank sample was determined as blank, and the RR for the contaminated sample was 89 ± 37 %. Additionally, for DON eight naturally contaminated wheat samples (as corn) were provided from Rosenmühle (Landshut, Germany). The concentration of DON was independently determined by the supplier using the aokinmycontrol® spectrometer FP470 and aokinmycontrol® DON kit (aokin AG, Berlin, Germany), which is a kinetic fluorescence polarization immunoassay. In all samples, DON was detected at concentrations from 347 to 1843 μg kg−1 (Table 3). Additionally, independent analyses were performed using an in-house (Chair of Hygiene and Technology of Milk) microtiter plate ELISA and by LC-MS (Agilent 1100 series LC/ 6300 SL ion trap MS, Agilent, Böblingen, Germany; Dr. T. Westermair, Dr. P. Walser, muva Kempten, Germany) using a U-[13C15]-deoxynivalenol internal standard from Biopure. At the Institute of Hydrochemistry, after milling for 30 s with a coffee mill (KM 13, R. Bosch GmbH, Stuttgart, Germany), the extraction was performed as described above. The estimated concentrations of DON which were obtained by different methods are summarized in Table 3. The data show that all methods yield comparable results, disregard some outliers. Whereas sample “2” was clearly determined as positive, i.e., DON concentration above the limit value, by all methods, the microarray underestimated sample “3” which was determined as positive by all others, obviously. Additional positive samples, i.e., exceedings of the set limit value, were found for samples “4” and “8” by aokinmycontrol® and ELISA, respectively. The observed differences are partially explainable, e.g., by the usage of different extractants which were selected according to the instructions of kit manufacturers or investigators settings. While the aokin method used water extraction followed by SPE clean-up, samples were extracted with methanol/water (60:40, v/v) for ELISA, i.e., water content was considerably higher and more efficient for extraction of DON than in the case of the multiplex immunoassay. On average, results of all immunochemical methods are more similar to each other than to LC-MS as is demonstrated by a between-method (interlaboratory) coefficient of variation <30 %. Especially, samples “2” “3” and “6” were overestimated by LC-MS which is difficult to explain. It needs further investigation, e.g., by usage of (C)RMs such as Trilogy® reference materials (www.r-biopharm.com). Even though the obtained results should not be overinterpreted due to the limited data set and differing number of measurements per sample by the analytical methods, the currently described approach offers the potential for a rapid and quantitative multianalyte analysis of mycotoxins in cereals. The obtained results are comparable to those of a recently published article which describes the use of the Bayer Quality Analyzer and Bayer Micro-Lab PWG system for multitoxin analysis (OTA, DON, T-2 toxin, and ZEA) in wheat [47]. The commercially available system is also based on an indirect competitive immunoassay but makes use of the planar waveguide technology, single-use biochips, and fluorescently labeled mycotoxin antibodies. With almost the same total analysis time (19 vs. 20 min) and similar working range for DON (76–1,173 vs. 400–3,000 μg kg−1), some lower recovery rate of 58–79 vs. 75–103 % was obtained in our study. The latter can be explained by the higher polarity of extractant mixture MeOH/water (70:30, v/v) used with the Bayer Quality Analyzer.
Conclusions
The parallel quantification of AFB1, OTA, FB1, and DON in three different cereal matrices with an automated chemiluminescence flow-through microarray was demonstrated. The total assay time, including extraction, extract dilution, measurement, and surface regeneration, was 19 min, and the crude extracts could be used without further purification other than filtration and dilution, allowing a rapid sample analysis. The prepared microarray chip was reusable for at least 50 times. Due to a batch-to-batch variation of the maximum signal intensity of more than 20 %, each new batch requires a new calibration of the analytical system demonstrating the need for further optimization of the chip preparation step. Cleanrooms with controlled environment are indispensable in this context. In-lab validation of the chip revealed that oat extract could be used as representative sample matrix for preparation of mycotoxin standards and determination of different types of cereals such as oat, wheat, rye, and maize at relevant concentrations set by the European Commission. The use of methanol/water (80:20, v/v) as extraction solvent yielded a good compromise to obtain acceptable recoveries of the four mycotoxins with distinct polarity features. This could be demonstrated with both fortified and naturally contaminated samples and commercially available reference materials. Some further improvement of the extraction yield might be expected by fine-tuning of the extractant mixture. Future efforts will be devoted to the inclusion of other mycotoxins relevant to cereals such like zearalenone and T-2/HT-2.
References
Shephard GS (2008) Chem Soc Rev 37:2468–2477
Urusov AE, Zherdev AV, Dzantiev BB (2010) Appl Biochem Microbiol 46:276–290
Prieto-Simon B, Karube I, Saiki H (2012) Food Chem 135:1323–1329
Li Y, Liu X, Lin Z (2012) Food Chem 132:1549–1554
Czeh A, Mandy F, Feher-Toth S, Torok L, Mike Z, Koszegi B, Lustyk GJ (2012) Immunol Methods 384:71–80
Bondarenko AP, Eremin SA (2012) J Anal Chem 67:790–794
Commission regulation (EC) No 1881/2006 (2006) Off J Eur Union 364:5–24
Commission regulation (EC) No 1126/2007 (2007) Off J Eur Union 255:14–17
IARC (1993) International Agency for Research in Cancer, Lyon, France. IARC 57:427–794
Köppen R, Koch M, Siegel D, Merkel S, Maul R, Nehls I (2010) Appl Microbiol Biotechnol 86:1595–1612
Kralj Cigic I, Prosen H (2009) Int J Mol Sci 10:62–115
Shephard GS, Berthiller F, Burdaspal PA, Crews C, Jonker MA, Krska R, MacDonald S, Malone RJ, Maragos C, Sabino M, Solfrizzo M, Van Egmond HP, Whitaker TB (2012) World Mycotox J 5:3–30
Hajslova J, Zachariasova M, Cajka T (2011) Mass spectrometry in food safety: methods and protocols. In: Zweigenbaum J (ed) Methods in molecular biology, 747th edn. Humana Press, New York, pp 233–258
Herebian D, Zühlke S, Lamshöft M, Spiteller M (2009) J Sep Sci 32:939–948
Sulyok M, Krska R, Schuhmacher R (2010) Food Chem 119:408–416
Monbaliu S, van Poucke C, Detavernier C, Dumoulin F, van de Velde M, Schoeters E, van Dyck S, Averkieva O, van Peteghem C, De Saeger S (2010) J Agric Food Chem 58:66–71
Krska R, Schubert-Ullrich P, Molinelli A, Sulyok M, MacDonald S, Crews C (2008) Food Addit Contam 25:152–163
Cichna-Markl M (2011) World Mycotox J 4:203–215
Cervino C, Asam S, Knopp D, Rychlik M, Niessner R (2008) J Agric Food Chem 56:1873–1879
Shephard GS et al (2011) World Mycotox J 4:3–2421
Lippolis V et al (2011) Anal Bioanal Chem 401:2561–2571
Li P, Zhang Q, Zhang W (2009) Trends Anal Chem 28:1115–1126
Liao J, Li H (2010) Microchim Acta 171:289–295
Tang D, Sauceda JC, Lin Z, Ott S, Basova E, Goryacheva I, Biselli S, Lin J, Niessner R, Knopp D (2009) Biosens Bioelectron 25:514–518
Basova EY, Goryacheva IY, Rusanova TY, Burmistrova NA, Dietrich R, Märtlbauer E, Detavernier C, Van Peteghem C, De Saeger S (2010) Anal Bioanal Chem 397:55–62
Beloglazova NV, Speranskaya ES, De Saeger S, Hens Z, Abé S, Goryacheva IY (2012) Anal Bioanal Chem 403:3013–2024
Lattanzio VMT, Nivarlet N, Lippolis V, Gatta DS, Huet A-C, Delahaut P, Granier B, Visconti A (2012) Anal Chim Acta 718:99–108
Zheng MZ, Richard JL, Binder J (2006) Mycopathologia 161:261–273
Li P, Zhang Z, Zhang Q, Zhang N, Zhang W, Ding X, Li R (2012) Electrophoresis 33:2253–2265
Njumbe Ediage E, Di Mavungu JD, Goryacheva IY, Van Peteghem C, De Saeger S (2012) Anal Bioanal Chem 403:265–278
Anfossi L, Baggiani C, Giovannoli C, D’Arco G, Giraudi G (2013) Anal Bioanal Chem 405:467–480
Kos G, Lohninger H, Krska R (2003) Anal Chem 75:1211–1217
Rasch C, Kumke M, Löhmannsröben HG (2010) Food Bioprocess Technol 3:908–916
Maragos CM (2009) World Mycotox J 2:221–238
Moises SS, Schäferling M (2009) Bioanal Rev 1:73–104
Tothill IE (2011) World Mycotox J 4:361–374
Peters J, Bienenmann-Ploum M, De Rijk T, Haasnoot W (2011) Mycotox Res 27:63–72
Dorokhin D, Haasnoot W, Franssen MCR, Zuilhof H, Nielen MWF (2011) Anal Bioanal Chem 400:3005–3011
Kloth K, Niessner R, Seidel M (2009) Biosen Bioelectron 24:2106–2112
Kloth K, Rye-Johnsen M, Didier A, Dietrich R, Märtlbauer E, Niessner R, Seidel M (2009) Analyst 134:1433–1439
Sauceda-Friebe J, Karsunke XYZ, Vazac S, Biselli S, Niessner R, Knopp D (2011) Anal Chim Acta 689:234–242
Cervino C, Weber E, Knopp D, Niessner R (2008) J Immunol Methods 329:184–193
Chu FS, Hsia MTS, Sun PS (1977) J Assoc Off Anal Chem Int 60:791–794
Usleber E, Märtlbauer E, Dietrich R, Terplan G (1991) J Agric Food Chem 39:2091–2095
Wolter A, Niessner R, Seidel M (2007) Anal Chem 79:4529–4537
Pope ME, Soste MV, Eyford BA, Anderson NL, Pearson TW (2009) J Immunol Methods 341:86–96
Tittlemier SA, Roscoe M, Drul D, Blagden R, Kobialka C, Chan J, Gaba D (2013) Mycotoxin Res 29:55–62
Acknowledgments
This research project was supported by the German Ministry of Economics and Technology (via AiF) and the FEI (Forschungskreis der Ernährungsindustrie e.V. Bonn); project AiF 381 ZN. Further, the authors thank Dr. T. Westermair and Dr. P. Walser (muva Kempten) for LC-MS analysis of deoxynivalenol in grain samples and Martina Kirsch, Rosenmühle Landshut (Germany) for providing naturally contaminated grain samples and performing analysis of deoxynivalenol by the aokinmycontrol® system. We are also grateful to Dr. G. Lystik (Soft Flow Biotechnology, Gödöllo, Hungary) for providing the anti-OTA monoclonal antibody and Huntsman Corporation for the free samples of DAPEG.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is dedicated to Professor Franz Dickert on the occasion of his 70th birthday.
Rights and permissions
About this article
Cite this article
Oswald, S., Karsunke, X.Y.Z., Dietrich, R. et al. Automated regenerable microarray-based immunoassay for rapid parallel quantification of mycotoxins in cereals. Anal Bioanal Chem 405, 6405–6415 (2013). https://doi.org/10.1007/s00216-013-6920-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-6920-3