
Abstract
Conventionally, microbial bioelectrochemical assays have been conducted using immobilized cells on an electrode that is placed in an electrochemical batch cell. In this paper, we describe a developed microfluidic platform with integrated microelectrode arrays for automated bioelectrochemical assays utilizing a new double mediator system to map redox metabolism and screen for genetic modifications in Saccharomyces cerevisiae cells. The function of this new double mediator system based on menadione and osmium redox polymer (PVI-Os) is demonstrated. “Wiring” of S. cerevisiae cells using PVI-Os shows a significant improvement of bioelectrochemical monitoring in a microfluidic environment and functions as an effective immobilization matrix for cells that are not strongly adherent. The function of the developed microfluidic platform is demonstrated using two strains of S. cerevisiae, ENY.WA and its deletion mutant EBY44, which lacks the enzyme phosphoglucose isomerase. The cellular responses to introduced glucose and fructose were recorded for the two S. cerevisiae strains, and the obtained results are compared with previously published work when using an electrochemical batch cell, indicating that microfluidic bioelectrochemical assays employing the menadione–PVI-Os double mediator system provides an effective means to conduct automated microbial assays.

Microfluidic platform for bioelectrochemical assays using osmium redox polymer “wired” living yeast cells
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microbial strain engineering is presently a common approach to improve biotechnological applications that comprise diverse areas, such as production of biomedicines [1], nutraceuticals [2], dairy products [3], beverages [4], biofuels [5], and precursors for organic synthesis [6] as well as bioremediation [7]. Although microbes can utilize a wide range of chemicals as energy source, strain engineering can further enhance this capability and improve the metabolic efficiency. Many of the key processes catalyzed by enzymes in microbes involve oxidation and reduction of chemicals. In enzyme-based bioelectrochemistry, the electron transfer between a substrate undergoing oxidation or reduction and the redox enzyme catalyzing the reaction is coupled to an electrode surface resulting in a current proportional to the rate of substrate oxidation or reduction. In an analogous manner, microbial bioelectrochemistry involves a coupling of the cellular redox processes to an electrode. The essential difference is that the enzymes are either in the plasma membrane or the intracellular environment.
The application areas of microbial bioelectrochemistry can be divided into research on microbial biosensors, metabolic mapping, screening of genetic modifications, and biofuel cells. All of these application areas rely on oxidation and reduction of different chemical species; however, from an operational perspective, certain fundamental differences can be seen. Biosensors map the microbial response to unknown environmental conditions to make a conclusion regarding the conditions the cells are exposed to. Analogously to enzyme biosensors, microbial biosensors have been used to detect a specific analyte, e.g., ethanol [8] and estrogen [9], offering advantages in terms of cost and time effectiveness by eliminating the necessity of enzyme purification as well as robustness in handling during biosensor fabrication and availability due to fast growth rates of microbes. They have been utilized also in environmental monitoring as an alternative to the conventional biochemical oxygen demand tests [10, 11] and for detection of herbicides [12] and aldehydes [13]. In metabolic mapping and screening of genetic modifications, the external conditions are controlled, and the microbial response is the unknown factor the experiments are intended to reveal. Proposed applications cover optimization of fermentation conditions [14, 15] and characterization of strain engineered microbes in terms of cytosolic and mitochondrial metabolism [16, 17] as well as intracellular enzyme activity [18]. Biofuel cells, on the other hand, utilize the redox processes for energy production with the aim to, e.g., convert organic waste to energy and power different electronic devices [19].
Communication between microbes and electrodes
Small molecules capable of accessing the active site of redox enzymes have been utilized widely to facilitate electron transfer between enzymes in cells and an electrode. If the active site of an enzyme is directed toward the extracellular environment, a hydrophilic compound, such as hexacyanoferrate (III), is a suitable electron transfer mediator [20]. In the case of enzymes having their active site toward the intracellular environment or entirely inside the cytosol, a lipophilic mediator, such as a quinone, capable of diffusing through the plasma membrane into the intracellular environment and back to the electrode is required [21]. In applications based on eukaryotic cells, such as Saccharomyces cerevisiae, a combination of two mediators (double mediator system), e.g., hexacyanoferrate (III) and menadione (a quinone), has been used [22, 23]. The lipophilic menadione can access the intracellular environment where it is reduced to menadiol, which then diffuses back to the extracellular environment to deliver its electrons to the hydrophilic hexacyanoferrate (III), reducing it to hexacyanoferrate (II). Reoxidation of hexacyanoferrate (II) at the electrode generates the current that serves as the indication of the intracellular redox activity. The use of the double mediator system improves the dynamics of the bioelectrochemical monitoring [23].
Although the usage of exogenously added electron transfer mediators has been a general means to couple cellular redox enzymes to electrodes in many application areas, research on microbial biofuel cells has given new insight into different ways microbes can establish electron transfer to electrodes without exogenous mediators [24]. Metal-reducing bacteria have been shown to form a direct contact with a solid electron acceptor, which can be the anode of a biofuel cell, using their outer membrane cytochromes. When the required direct contact with the growth substrate is not possible, the microbes can alternatively “wire” themselves to the growth substrate using conductive pili that are coupled to the membrane-bound cytochromes. Additionally, certain microbes are also able to excrete secondary metabolites, which are small molecular electron transfer mediators. Although the ability of a biofilm to establish its own electron transfer is now widely utilized in constructing biofuel cells, the approach limits the choice of microbes and requires a certain time before the electron transfer is fully established.
When using microbes that are not able to form their own electron transfer to a growth surface, an alternative approach is to establish electron transfer between enzymes in microbes and an electrode surface by using redox polymer “wiring.” Aside from facilitating applications with a larger number of microbes, this approach also allows fast bioelectrochemical screening of different microbes without the need to have a biofilm formed by the microbes. Osmium redox polymers, which originally were introduced by A. Heller and coworkers for enzyme-based applications [25], have been successfully used to “wire” different bacteria, such as Gluconobacter oxydans [26], Pseudomonas putida, Pseudomonas fluorescens [27], Escherichia coli [28], Bacillus subtilis [29], and Rhodobacter capsulatus [30], as well as the yeast Hansenula polymorpha [31].
Miniaturized systems for microbial bioelectrochemistry
Miniaturization of systems that can be used to study microbes has opened new possibilities and has been adopted in many applications [32]. Generally, a significant development toward miniaturization of systems suitable for the study of living cells concerns development of microfluidic systems. When such studies are combined with bioelectrochemistry, an additional area of miniaturization is related to design and fabrication of microelectrodes that can be interfaced to the systems. However, in the field of microbial bioelectrochemistry, the miniaturization of systems, especially in the form of microfluidic systems, is still at an early stage. The main development of miniaturized bioelectrochemical systems has been conducted in the field of microbial biofuel cells, which is indicated by some recent reviews [33–35]. Microfluidic systems coupled with electrodes can offer possibilities in terms of an increased surface area-to-volume ratio, which in a biofuel cell can improve the ability of effective biofilm formation and give a more rigorous control over the microenvironment around the microbes. Additionally, miniaturization and parallelization of electrode systems have been shown to open possibilities to screen microbial consortia to find suitable microbes for constructing biofuel cells [36]. In other areas of microbial bioelectrochemistry, miniaturization has primarily concerned design and fabrication of electrode arrays for evaluation of the metabolic status of microbes [16, 18, 23, 37]. A microfluidic system with integrated electrode arrays has been devised for the study of biofilm formation of Candida albicans, utilizing bioelectrochemistry for evaluation of respiration activity [38].
In this paper, we describe a microfluidic platform with integrated electrode arrays that can be used to evaluate microbial redox metabolic status and screen for genetic modifications that are utilized in strain engineering to alter microbial redox metabolism. The application of the platform facilitates fast assays of microbes without any necessity of initial biofilm formation. The function of the platform is demonstrated using S. cerevisiae cells. As a significant part of the system development, we demonstrate osmium redox polymer “wiring” of the cells to the electrode arrays combined with the application of the lipophilic mediator menadione. This new double mediator system shows great potentials in microfluidic applications with cells that are not strongly adherent, since the 3D network created by the osmium redox polymer functions as an immobilization matrix.
Materials and methods
Chemicals and materials
Menadione (2-methyl-1,4-naphthoquinone) (USP), d-(+)-glucose (BioXtra), d-(−)-fructose (BioXtra), potassium chloride (BioXtra), monobasic potassium phosphate (cell culture tested), potassium hydroxide (semiconductor grade), hydrogen peroxide (USP), potassium hexacyanoferrate (II) trihydrate (BioUltra), potassium hexacyanoferrate (III) (BioUltra), ethanol (96 %), polyethylenimine (PEI) (branched, average M w ∼25,000 Da), tryptone, and yeast extract were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA). Osmium redox polymer, poly(1-vinylimidazole)12-[Os-(4,4′-dimethyl-2,2′-bipyridyl)2Cl2]+/2+ (PVI-Os), was a gift from TheraSense, Inc. (Alameda, CA, USA). All solutions were prepared using ultrapure water (resistivity 18.2 MΩ cm) from a Milli-Q® water purification system (Millipore Corporation, Billerica, MA, USA). AZ® 5214E positive photoresist (MicroChemicals GmbH, Ulm, Germany) and SU-8 2005 negative photoresist (MicroChem Corp., Newton, MA, USA) were used in lithographic fabrication of microelectrode array chips. Other chemicals used in lithography were chosen according to the recommendation of the photoresist suppliers. Polycarbonate (PC) (Bayer MaterialScience AG, Leverkusen, Germany), poly(methyl methacrylate) (PMMA) (Röchling Technische Teile KG, Mainburg, Germany), and silicon adhesive (INT TA106) (Intertronics, Oxfordshire, UK) were used for fabrication and interfacing of microfluidic components. Electric connections were made of spring-loaded pin arrays (811-S1-NNN-10-015101) (Preci-Dip SA, Delémont, Switzerland). Silver wire (Ø 500 μm) (Goodfellow Cambridge Limited, Huntington, UK) was used for fabricating pseudo-reference electrodes (pREs).
Instrumentation
Optical density at 620 nm (OD620) was determined using a U-1100 spectrophotometer (Hitachi Ltd., Tokyo, Japan). Yeast cells were cultured in an I26 incubator shaker (New Brunswick Scientific, Enfield, CT, USA) and centrifuged using an Eppendorf 5810 centrifuge (AG, Hamburg, Germany). Microscopic imaging was done using an Axio Imager M1m microscope equipped with an AxioCam MRc5 computer-controlled CCD camera (Carl Zeiss AG, Göttingen, Germany). Cyclic voltammetric, square wave voltammetric, and amperometric experiments were performed using a computer-controlled eight-channel CHI1010A multipotentiostat (CH Instruments, Inc., Austin, TX, USA). Micropumps were operated with Lego® Interactive Servo Motors (Lego System A/S, Billund, Denmark) that were controlled by an NXT Intelligent Brick (Lego System A/S, Billund, Denmark) executing a LabView-based script. Micromilling was done using a Mini-Mill/3Pro system (Minitech Machinery Corporation, Norcross, GA, USA) executing G-code generated by EZ-CAM15 Express software (EZCAM Solutions, Inc., New York, NY, USA). UV activation, bonding, and laser cutting were done with a 5000-EC Series UV Curing Flood Lamp System (Dymax Corporation, Torrington, CT, USA), a PW20 hydraulic press (Paul-Otto Weber GmbH, Remshalden, Germany), and a 48-series CO2 laser system (Synrad, Inc., Mukilteo, WA, USA), respectively. O2 plasma treatment was done using a Tepla 300 microwave plasma processor (PVA Tepla America, Corona, CA, USA). UV lithographic processes in the clean room (Danchip, Technical University of Denmark) employed standard equipment.
System design and fabrication
Microelectrode array chips were fabricated through standard lithographic processes on wet-oxidized 500-μm-thick 4-in. silicon wafers (one side polished). The electrode structures, leads, and contact pads were lithographically defined in 1.5-μm-thick spin-coated positive photoresist using an image reversal process [39]. After photoresist development, 20 nm of titanium (adhesion layer) and 200 nm of gold were deposited on the wafers through electron beam evaporation. The process was finalized with lift-off in acetone under ultrasonication. To obtain defined exposed electrode areas and contact pads, the wafers were spin coated with a 5-μm-thick layer of SU-8, which was subsequently lithographically patterned. Residual SU-8 in areas that were to be exposed was removed using a gentle O2 plasma treatment. The fully processed wafers were diced to obtain individual microelectrode array chips.
The microfluidic motherboard, housing a peristaltic micropump, microfluidic reservoir chips (reagents and waste), and a microfluidic chip, was fabricated of 4 mm PC. The components of the reagent reservoirs and disposable microfluidic chips were fabricated of 3 and 1 mm PMMA, respectively. The machining of all the plastic components was done by micromilling.
The individual micromilled PMMA components of a microfluidic chip were bonded together using 1-min UV activation of the surfaces followed by a heat/pressure-assisted bonding (20 min at 85 °C/4.4 MPa). Prior to UV exposure, the PMMA sheets were gently cleaned with 96 % ethanol and ultrasonicated for 5 min in Milli-Q water. Each bonded microfluidic chip was interfaced to an electrode array microchip with a 50-μm-thick silicon adhesive layer which was laser cut.
After fabrication of a microfluidic chip and interfacing it to a microelectrode array chip, an Ag|AgCl pRE was attached to the outlet side of each microfluidic chamber (for preparation, characterization, and positioning, see Section 1 in the Electronic supplementary material).
Microelectrode array preparation and characterization
Each microelectrode array chip was tested thoroughly prior to interfacing it with a microfluidic chip. The testing was done in a PMMA holder, which forms an individual vial around each electrode array (see Section 4 and Fig. S4 in the Electronic supplementary material for details). Prior to electrochemical testing, the electrodes were cleaned for 10 min with the mixture of H2O2 (25 % v/v) and KOH (50 mM) followed by a potential sweep of the working electrodes (WEs) from −200 to −1,200 mV (vs. pRE) at 100 mV/s in 50 mM KOH as previously described [40]. After cleaning, the WEs were tested by cyclic voltammetry in 10 mM potassium hexacyanoferrate (II) and (III) prepared in 0.1 M KCl. The electrochemical part of the cleaning and the subsequent testing can be done using either the on-chip gold reference electrode (RE) and counter electrode (CE) or a set of miniaturized homemade Ag|AgCl pRE and platinum CE that can be placed with a manual micromanipulator into the vials of the PMMA holder. Modification of each working electrode with PVI-Os can be done immediately after testing or alternatively after interfacing the electrode chip with a microfluidic chip and assembling on the microfluidic motherboard. PVI-Os was electrodeposited based on a previously described procedure [41] from a 10-mg/ml solution of the polymer in 0.1 M KCl by applying square potential waves between 800 and −300 mV (vs. Ag|AgCl pRE) (50 potential step cycles with a 2-s residence time at each potential).
Growth of S. cerevisiae strains
Two strains of S. cerevisiae were used in the experiments: the parental strain ENY.WA-1A (MATα, ura3-52, leu2-3,112, trpl-289, his3-Δ1, MAL2-8c, MAL3, SUC3) [42] and its deletion mutant strain EBY44 (ENY.WA-1A pgi1-1Δ::URA3) [43]. Both strains were gifts from Prof. E. Boles (Institute of Microbiology, Frankfurt, Germany). Pre-culturing of cells was done overnight at 30 °C in 50-ml shake flasks having 5 ml of medium (20 g/l tryptone, 10 g/l yeast extract, 20 g/l d-(-)-fructose and 1 g/l d-(+)-glucose in 100 mM potassium phosphate buffer at pH 6.2 (pH adjusted using KOH)) and shaken at 180 rpm. After pre-culturing, the cells were harvested by centrifugation at 3,500 rpm at room temperature (RT) for 5 min. The obtained pellet was then diluted with culture medium to OD620 of 0.5 and used to inoculate 100 ml of culture medium in 500-ml shake flasks. The cells were grown as stated above until the early stationary phase and immediately used for the experiments.
Cell preparation and immobilization
After harvesting, the cells were rinsed twice with phosphate-buffered saline (PBS) (10 mM potassium phosphate and 100 mM KCl; pH 7.2 adjusted using KOH) and resuspended in PBS. The OD620 was adjusted initially to 70 as previously described [44]. One milliliter of the cell suspension was mixed with 1 ml of PBS containing 2 mg/ml PEI (final OD620 of 35). The cells were incubated for 1 h at RT under gentle shaking. The PEI-coated cells were diluted with PBS to OD620 of 0.1, and 950 μl of the cell suspension was mixed with 50 μl of PVI-Os (10 mg/ml diluted in PBS) resulting in a suspension having ca. 5 × 106 cells/ml based on previously published results [16]. The outlet reservoirs on the microfluidic platform were filled with cell suspension (two reservoirs for each genotype), and the platform was placed on the microscope stage for observation during cell seeding. The cell suspensions were fed into the chambers of the microfluidic chip by reverse pumping at the flow rate of 5 μl/min. When the cell suspension had completely traversed the area of the microfluidic chambers, the pumping was stopped and the cells were allowed to sediment for 1 h. The excess of cell suspension was removed from the outlet reservoirs.
Bioelectrochemical assay of cellular redox activity
After 1-h sedimentation, the cellular assay was initiated by filling the microfluidic chambers through forward pumping (10 μl/min flow rate) with PBS containing 10 mM glucose or fructose (incubation buffer). This solution also flushed away the PEI/PVI-Os containing PBS from the cell immobilization step. The perfusion with incubation buffer was continued for 15 min. Ca. 5 min after starting the perfusion, the WEs were poised at 400 mV vs. Ag|AgCl pRE to start recording of the baseline. After the completed perfusion with incubation buffer, the solution was changed to one also containing 100 μM menadione (assay buffer), prepared according to a previously described procedure [44]. The perfusion with assay buffer was continued at the flow rate of 10 μl/min while the current response generated by the cells was recorded. The bioelectrochemical current responses were recorded simultaneously in four microfluidic chambers (two chambers for ENY.WA-1A strain and two chambers for EBY44 strain). Each S. cerevisiae strain was exposed to glucose and fructose containing incubation and assay buffer. In each microfluidic chamber, two WEs were included for current recordings, generating simultaneously eight current–time traces. The results are reported as averages of current–time traces recorded on two WEs.
Results and discussion
Microfluidic platform with integrated electrode arrays
The aim in designing the presented microfluidic platform was to facilitate easy handling of assays for bioelectrochemical probing of nonadherent S. cerevisiae cells. The platform is composed of a generic reusable motherboard with an eight-channel peristaltic micropump (for details, see Section 2 and Fig. S2 in the Electronic supplementary material) and microfluidic reservoir chips for reagents and waste collection during assays as well as an exchangeable microfluidic chip having integrated electrode arrays that can be mounted on the motherboard to form the complete platform. The overall structure of the platform is shown in Fig. 1.

An assembled microfluidic platform comprising a reusable motherboard and a disposable microfluidic chip with an integrated microelectrode array chip
The peristaltic micropump is operated using a Lego® step motor, which is attached to a shaft in the middle of the pump. The fluidic operations during experiments were automated using a preprogrammed LabView-based script stored in the memory of the Lego® NXT Intelligent Brick. The executed script allowed independent functions for priming the microfluidic chip, preparation of the electrodes, seeding of S. cerevisiae cells, and performing the bioelectrochemical assays. The general testing and characterization of the pumping uniformity and flow rates of the eight-channel micropumps have been published earlier [45]. The fluidic operations in the fabricated microfluidic platform were calibrated with accurate flow rates obtained between 25 nl/min and 85 μl/min by adjusting the rotation speed of the step motor. The pumping capacity per rotation was 650 ± 8 nl (±standard error of the mean; n = 24), determined by three repetitions on each of the eight channels.
Figure 2a shows an enlargement of a bonded two-layer microfluidic chip having four chambers for loading of S. cerevisiae cells. Since the utilized interconnection blocks and PDMS ribbons were designed for eight channels, the microfluidic chips have four short dummy channels with openings to eliminate pressure buildup during operation of the pump. Due to the 180° symmetry of microfluidic chip, no polarity needed to be considered when integrating it with the microelectrode arrays or microfluidic motherboard. Each inlet and outlet channel is 500 μm (width) × 250 μm (depth) connecting a chamber through a widening part that ends with a semicircular fall (schematically shown in the inset of Fig. 2b) at each end of the chamber in which the cells are loaded and measurements are conducted. The dimensions of each chamber are length 13 mm, width 4.5 mm, and height 750 μm. The construction of the microfluidic device is completed by integration of the bonded microfluidic chip with a microelectrode array chip.

a A bonded microfluidic PMMA chip and b a schematic explosion view of the assembly of a microfluidic chip and its integration with a microelectrode array chip (inset a schematic view of a semicircular fall at the end of the widening part of an inlet channel)
Figure 2b shows a schematic explosion view of the assembled microfluidic chip together with the integrated microelectrode array chip, as well as the spring-loaded pin arrays that form the electric contacts to the potentiostat (the silicon adhesive gasket that is used to integrate the microfluidic chip and microelectrode array chip is not shown in this schematic view). The inset of Fig. 2b shows a schematic view of the widening part of a channel ending with the semicircular fall. Section 1 and Fig. S1 in the Electronic supplementary material show the positioning of the pRE attached through a hole on the outlet side of the microfluidic chamber. The closed microfluidic chambers with the semicircular ends were designed to provide a laterally even flow to ensure reliable performance during the cell seeding and measurements. Section 3 and Fig. S3 in the Electronic supplementary material show a finite element simulation indicating the flow uniformity.
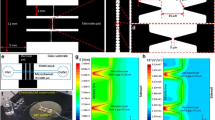
Figure 3a shows a microelectrode array chip with four measurement sites each comprising three WEs, having five microbands (25 × 1,000 μm; see the inset of Fig. 3a) formed by lithographic processing of the SU-8 layer, passivating the areas of the chip where the gold should not be exposed to the electrolyte solution. The 5-μm-thick SU-8 layer forms an elongated “vial” around each microband (a schematic cross-sectional view shown in Fig. 3b), which helps in loading a monolayer of S. cerevisiae cells having the approximate diameter of 5 μm. The microelectrode array chips also have an in-built CE and RE that are common for each of the measuring sites. However, in the application described in this paper, the in-built gold RE was replaced by the above-described Ag|AgCl pRE, which provides a more accurate and long-term stable control over the applied potential (for characterization of the pREs, see Section 1 in the Electronic supplementary material).

a A magnified image of a microelectrode array chip with four measuring sites each having three individually addressable WEs as well as a common counter electrode and reference electrode (inset a magnified image of a WE with five 25 × 1,000-μm microbands). b A schematic view of the microelectrode array chip structure showing a cross section of three microbands
In previous bioelectrochemical studies of S. cerevisiae cells, microband electrodes, having the dimension of 25 × 1,000 μm, have been utilized successfully in an electrochemical batch cell for recording current responses from cells immobilized in Ca-alginate gel [16, 18, 23, 44]. Based on characterization, a Ca-alginate gel layer on an electrode surface is approximately 100 μm thick [16], facilitating a sufficiently large number of cells to be immobilized in the volume element defined by the active electrode area. In the microfluidic application described in this paper, a monolayer of cells is immobilized on the WEs. To obtain a sufficiently large number of cells on each WE, five microbands were lithographically defined on each WE. The usage of microband electrodes instead of the entire gold area, which would significantly increase the number of immobilized cells, is furthermore significant in order to decrease the capacitive effects brought about by an increased electrode area.
Electrode array preparation
Prior to assembling the microfluidic chip with the integrated silicon chip having microelectrode arrays, each silicon chip was tested in an electrochemical batch setup to ensure a proper electrochemical function of the electrode arrays (for details of the testing setup, see Section 4 and Fig. S4 in the Electronic supplementary material). The testing comprised both cleaning of the electrodes and cyclic voltammetric characterization using a 10-mM solution of hexacyanoferrate (II) and (III). The cleaning procedure ensures that the electrodes function reproducibly, which is a prerequisite for accurate bioelectrochemical assays where only the biological material should be the entity resulting in variation in the recorded current. The effect of the cleaning process for the 12 WEs of a microelectrode array chip can be seen in Fig. S5 in the Electronic supplementary material. The excellent reproducibility of the electrochemical behavior obtained for the electrode array guarantees a successful implementation of detection protocols when using the electrode arrays.
To ensure an effective electrochemical communication between the WE surface and the S. cerevisiae cells, the WEs were modified using electrodeposition of the PVI-Os redox polymer (see the structural representation in Fig. 4a). This approach has been proposed previously as a suitable means to co-deposit enzymes to facilitate their “wiring” on electrode surfaces [41]. It has found applications in construction of both enzyme-based biosensors [46] and biofuel cells [47]. In such applications, the Os redox polymer concentrations have been equal to or below 1 mg/ml, facilitating a sufficient co-deposition of enzymes. However, in the experiments described in this paper, 1 mg/ml of Os redox polymer during electrodeposition (50-cycle square potential wave) resulted in low current responses in the bioelectrochemical assays. Independent of the used cell densities, the obtained current responses were clearly below 20 nA. To provide sufficiently high current responses during assays without increasing the deposition time, the concentration of Os redox polymer was increased to 10 mg/ml. This increased the availability of the Os redox polymer at the electrode interface during deposition, yielding a layer that provided an increase in the bioelectrochemical response to the level shown in the “Bioelectrochemical assay of cellular redox activity” section. The obtained result indicates that the electrodeposited Os redox polymer serves as a foundation layer for the subsequent deposition of “wired” S. cerevisiae cells (described below). The cyclic voltammograms in Fig. 4b indicate the increased electrochemical response of the electrodeposited PVI-Os in comparison with the corresponding response of PVI-Os in solution.

a Structure of osmium redox polymer (PVI-Os). b Cyclic voltammograms of PVI-Os in solution (10 mg/ml in 0.1 M KCl) and after electrodeposition (scan rate 50 mV/s; potentials vs. pseudo-Ag|AgCl RE)
The microfluidic platform provides a versatile performance, allowing electrode modification online. The necessary reagents can be accommodated in the vials of the microfluidic reservoir chip (Fig. 1), and the electrochemical processes can be performed upon filling the chambers of the microfluidic chip, integrated with the microelectrode array chip.
PVI-Os “wiring” and microfluidic seeding of S. cerevisiae cells
The previously published applications on “wiring” of microbial cells to electrode surface using Os redox polymers have utilized either gold electrodes [26, 27, 29, 30] or graphite electrodes [28, 30, 31]. The common approach has been to pipette a small volume (3–5 μl) of an aqueous polymer solution on the electrode surface and allow it to dry prior to placing a small volume of a cell suspension on the dried Os polymer layer. To retain the introduced cell layer on the electrode during a bioelectrochemical assay, the electrodes have been covered using a dialysis membrane [26–30]. In the case of using H. polymorpha yeast cells, the dried cell layer was additionally covered with a small volume of Os polymer solution [31]. A compact Os polymer network around the cell layer was achieved by using a short exposure to a buffer with either lowered or elevated pH, which caused precipitation of the utilized Os polymer. In this application, the cell layer could be retained on the electrode surface without covering it with a dialysis membrane.
In the microfluidic application presented in this paper, the S. cerevisiae cells were “wired” with Os redox polymer using an approach that was suitable for microfluidic cell seeding into the chambers of the microfluidic chip. To facilitate an effective formation of a 3D network of Os polymer around the seeded cells, these were initially incubated off-line with PEI, which on virtue of its positive net charge forms an electrostatically retained layer on the cells. After introduction of Os polymer to the PEI-coated yeast cells, these were placed in the reservoirs of the outlet side of the microfluidic platform for cell seeding by reverse pumping. The suspension of Os polymer “wired” cells was sufficiently low in viscosity to allow easy and reproducible handling during microfluidic cell seeding.
When using microfluidics, the density of seeded cells on the electrodes can be optimized to a desired level by adjusting the initial density of the cell suspension as well as the flow rate and duration of pumping. However, cell seeding during continued pumping results in cell deposition in the microfluidic channels upstream of the chambers. In order to minimize this problem, pumping was stopped when the cells had traversed through the chambers. To obtain a sufficient cell density on the electrodes without a continued pumping, the density of the cell suspension was adjusted to 5 × 106 cells/ml. Since the chambers of the microfluidic chip are transparent, microscopic observation during cell seeding and counting of the seeded cells are possible. Using the adopted cell seeding approach, the obtained average cell density per WE (averaged for four microfluidic chambers each having three WEs) was approximately 2,900 cells with an RSD of 2.6 %. Figure 5a shows deposited S. cerevisiae cells on a portion of two microbands of a WE. The same accuracy in cell seeding can be achieved from device to device based on optimized component fabrication and system assembly.

a A magnified image of S. cerevisiae cells deposited on a microelectrode array chip (a portion of two microbands are shown). b A schematic representation of S. cerevisiae cell “wiring” with PVI-Os to the working electrode surface (modified with electrodeposited PVI-Os) and the functional principle of the double mediator system menadione–PVI-Os (see text for detailed clarification) (M menadione, MH 2 menadiol, MRE menadione-reducing enzymes, m metabolic pathways generating NAD(P)H)
When mapping cellular metabolism and screening genetic modifications, “wiring” of cells with Os redox polymer is not sufficient alone to contribute to a bioelectrochemical response unlike in applications relying on membrane-bound enzymes in bacteria [26–30] or yeast [31]. Since it is necessary to establish communication with intracellular enzymes, a double mediator system combining a lipophilic and hydrophilic mediator is significant, as has been demonstrated previously [16–18, 22, 23, 44]. Figure 5b shows a schematic view of S. cerevisiae cells wired with Os redox polymer, as well as the functional principle of this new double mediator system, combining the Os redox polymer with the lipophilic mediator menadione. As a lipophilic mediator, menadione is able to penetrate through the plasma membrane (denoted as the primary mediator) to communicate with intracellular menadione-reducing enzymes [23], including the mitochondrial complexes [16, 17]. Due to its lipophilicity, the reduced form of menadione, menadiol (MH2), is able to diffuse out of the cells. Based on the functional principle of the double mediator system, menadiol is reoxidized extracellularly by the Os redox polymer mediator (denoted as the secondary mediator). The reduced Os redox centers deliver the electrons to the electrode, giving rise to a current that is proportional to the intracellular redox activity.
In previously published work, the secondary mediator has been hexacyanoferrate (III), which is reduced to hexacyanoferrate (II), capable of delivering electrons to the electrode [16, 18, 23, 44]. In an application utilizing S. cerevisiae cells immobilized in a Ca-alginate matrix on electrodes and relying on vigorous magnetic stirring, the obtained current response was shown to be about 108 nA at the steady state [16]. In a microfluidic application of the hexacyanoferrate (III)–menadione double mediator system, the soluble hexacyanoferrate (III) was shown to reoxidize the formed menadiol; however, the obtained steady-state current was only about 14 nA [48]. This significantly lower steady-state current obtained in the microfluidic application, despite a comparable cell density, is caused by the fact that a considerable fraction of the reduced soluble secondary mediator is flushed out of the microfluidic chamber without being reoxidized on the electrode. Hence, the consequence is that the observed current is lower than expected based on the cellular redox activity.
When “wiring” S. cerevisiae cells with the secondary mediator Os redox polymer, forming a 3D network around each cell, the menadiol that diffuses out of the cells encounters the Os redox polymer, which is in redox contact with the electrode surface modified with electrodeposited Os polymer. This new double mediator system is capable of efficiently monitoring the dynamic changes in cellular redox activity, accurately relating the recorded current in real time to the metabolic processes that either generate or consume the cellular redox equivalents (cofactors), such as NADH and NADPH. Hence, “wiring” of S. cerevisiae cells using Os redox polymer in combination with menadione is novel and significant for optimal microfluidic bioelectrochemical assays as is demonstrated by the current responses presented below.
Aside from the enhanced electric contact between the cells and the electrode surface, the Os polymer in combination with PEI functions as an effective 3D immobilization matrix for retaining the cells on the electrodes in the microfluidic system. Furthermore, unlike other immobilization matrices, such as Ca-alginate gel that has been used in bioelectrochemical applications in electrochemical batch cells having vigorous stirring [16, 18, 23, 44], the obtained bioelectrochemical responses indicate that the 3D network of PEI and Os polymer does not form any significant mass transfer barrier. This ensures that the introduced cellular effectors are readily transported to the cells, facilitating a cellular response, which is essentially only dependent on the biological factors, such as diffusion or active transport through the cell membrane and intracellular enzyme activities. However, if the density of seeded cells is increased to comprise multiple layers of cells, the cells themselves form a mass transfer barrier, retarding the access of menadione to the cells closest to the electrode surface. This can significantly increase the time required for obtaining a steady-state current (for details, see Section 6 and Fig. S6 in the Electronic supplementary material).
Bioelectrochemical assay of cellular redox activity
In bioelectrochemical monitoring of cellular redox activity using a double mediator system, the mediators are chosen based on the requirements posed by the reduction potentials of the intracellular cofactors and the redox enzymes utilizing them. This has been schematically presented by Spégel et al. [23], indicating the suitability of menadione to probe intracellular redox activity on virtue of its less negative reduction potential in comparison with the values characteristic of cellular cofactors. Furthermore, hexacyanoferrate (III) has a reduction potential, which is more positive than that of menadione, making it capable of readily reoxidizing menadiol. In terms of the menadione–PVI-Os double mediator system, presented in this paper, this matching of reduction potentials had to be taken into consideration in the choice of the Os redox polymer. Generally, the reduction potential of Os polymers can be fine tuned to suit a specific application [49], which has been demonstrated in “wiring” of H. polymorpha cells [31]. In the application presented here, the chosen Os polymer poly(1-vinylimidazole)12-[Os-(4,4′-dimethyl-2,2′-bipyridyl)2Cl2]+/2+, having a reduction potential of approx. +150 mV vs. Ag|AgCl [29], is suitable as the secondary mediator to deliver the electrons originating from the intracellular redox processes and shuttled out of the cells by menadiol (for details of menadione electrochemistry in the presence of Os redox polymer, see Section 7 and Fig. S7 in the Electronic supplementary material).
To illustrate the capability of the menadione–PVI-Os double mediator microfluidic system in accurately monitoring the dynamics of intracellular redox activity, EBY44 [43] and its parental strain ENY.WA [42] were chosen as model strains. Since the bioelectrochemical response of these strains has been characterized earlier using the double mediator system hexacyanoferrate (III)–menadione, it is easy to compare the performance of PVI-Os as a new proposed secondary mediator. In the cytosol of S. cerevisiae cells, different monosaccharides, such as glucose and fructose, are metabolized in the two main metabolic pathways, the pentose phosphate pathway (PPP) generating NADPH and the glycolytic pathway (GP) generating NADH. The nonpreferential distribution of glucose and fructose between these two metabolic pathways is caused by the activity of the enzyme phosphoglucose isomerase (PGI), which is the branching point between these two pathways. This is the case in the parental strain ENY.WA (see Fig. 6a, right panel). In the EBY44 strain, the gene encoding for PGI has been deleted resulting in a differential distribution of glucose and fructose. Thus, here, glucose is primarily metabolized in the PPP generating NADPH, and fructose is metabolized in the GP generating NADH (see Fig. 6b, right panel).

Averaged bioelectrochemical response (each recorded on two WEs) of the a ENY.WA (left panel) and b EBY44 (left panel) strains of S. cerevisiae to glucose and fructose using the double mediator system menadione–PVI-Os (arrows indicate the time of menadione addition). Cytosolic metabolic pathways of a ENY.WA (right panel) and b EBY44 (right panel) (see text for detailed clarification) (PPP pentose phosphate pathway, GP glycolytic pathway, PGI phosphoglucose isomerase enzyme)
Figure 6a and b (left panels) show averaged current–time traces, each recorded on two WEs, during the bioelectrochemical probing of the metabolism of ENY.WA and EBY44 cells, respectively, when these metabolize glucose and fructose. In both cases, the generation of NADPH or NADH increases the recorded current as a consequence of the increased activity of the menadione-reducing enzymes, which utilize these cofactors. In the case of the ENY.WA cells, the obtained average steady-state response upon introduction of glucose and fructose is 71 ± 2 and 70 ± 2 nA, respectively. Based on t test (95 % level), the responses are equal (p = 0.653) as is expected based on the distribution of glucose and fructose in the metabolic pathways (Fig. 6a, b (right panels)). On the other hand, in the case of the EBY44 cells, introduction of glucose results in the average current response of 128 ± 4 nA, which is 2.4 times higher than that generated upon introduction of fructose (54 ± 3 nA). This significant difference can be explained by the fact that glucose metabolism predominantly generates NADPH, which has been shown to be the preferred cofactor utilized by menadione-reducing enzymes [23].
The bioelectrochemical responses shown in Fig. 6 were obtained in an automated assay in the microfluidic platform, where ENY.WA cells were seeded in two chambers and EBY44 cells in the two other chambers and each strain subjected to a flow of glucose or fructose containing incubation and assay buffer. Aside from showing that the microfluidic assay using the double mediator system menadione–PVI-Os can generate responses that are biologically relevant, the results also indicate that the level of recorded current corresponds to previously reported results using cells of the same S. cerevisiae strains immobilized in Ca-alginate gel on microelectrodes [16]. The presented results were obtained using the cell seeding approach described above, yielding about 2,900 cells per WE. However, to successfully perform assays in the designed microfluidic platform, the precise number of seeded cells does not need to be specifically optimized. The primary requirements are that the chosen cell density yields a sufficiently high current response (for the effect of multiple cell layers on the obtained response, see Section 6 in the Electronic supplementary material), and the number of cells in a set of experiments is constant to facilitate statistically relevant comparison between responses.
The demonstrated assay clearly shows that microbial bioelectrochemistry can be automated using microfluidics and that “wiring” of S. cerevisiae cells with Os redox polymer is a novel way to enhance the response in a double mediator system applied in a microfluidic environment since the electrons delivered by the lipophilic mediator can be more effectively detected due to the 3D network of the Os polymer. Although the presented model microfluidic platform utilized four chambers, the same approach can be parallelized easily by designing and fabricating a microfluidic chip and microelectrode array chip with more chambers and measuring sites, respectively. The same reusable microfluidic motherboard with an eight-channel micropump can be automated for more parallelized assays. Furthermore, the motherboard can accommodate an additional multichannel micropump or a programmable valve [50] for improved assay automation.
Conclusions
Bioelectrochemical screening of dynamic changes in redox metabolism of S. cerevisiae and other microbial cells as a consequence of genetic modifications has been shown in previous publications. Such assays have been conducted using electrode systems and immobilization techniques that are suitable for electrochemical batch cells. This paper presents the development of a microfluidic platform and its application for automated bioelectrochemical assays for screening genetic modifications in S. cerevisiae cells. The platform comprises a reusable motherboard having a peristaltic micropump and reservoirs for reagents and waste, as well as an exchangeable microfluidic chip integrated with a microelectrode array chip. In combination with a double mediator system based on the soluble lipophilic menadione and an osmium redox polymer that effectively “wires” the cells to the electrode surface, the presented microfluidic approach improves the possibilities of bioelectrochemical assays toward automation and higher throughput. The system is here demonstrated for screening of genetic modifications, but can be adopted easily for many other types of cellular assay, such as whole-cell biosensors, drug screening, and functional cellular assays. Furthermore, the motherboard facilitates a further parallelization by changing the design of the disposable microfluidic chip and the integrated microelectrode array chip.
References
Kjeldsen T, Balschmidt P, Diers I, Hach M, Kaarsholm NC, Ludvigsen S (2001) Biotechnol Gen Eng Rev 18:89–121
Hugenholtz J, Smid EJ (2002) Curr Opin Biotechnol 13:497–507
Hugenholtz J (2008) Int Dairy J 18:466–475
Nevoigt E, Pilger R, Mast-Gerlach E, Schmidt U, Freihammer S, Eschenbrenner M, Garbe L, Stahl U (2002) FEMS Yeast Res 2:225–232
Zhang F, Rodriguez S, Keasling JD (2011) Curr Opin Biotechnol 22:775–783
Stewart JD (2000) Curr Opin Biotechnol 11:363–368
Pieper DH, Reineke W (2000) Curr Opin Biotechnol 11:262–270
Ikeda T, Kato K, Maeda M, Tatsumi H, Kano K, Matsushita K (1997) J Electroanal Chem 430:197–204
Baronian KHR, Gurazada S (2007) Biosens Bioelectron 22:2493–2499
Nakamura H, Suzuki K, Ishikuro H, Kinoshita S, Koizumi R, Okuma S, Gotoh M, Karube I (2007) Talanta 72:210–216
Pasco N, Baronian K, Jeffries C, Hay J (2000) Appl Microbiol Biotechnol 53:613–618
Rawson DM, Willmer AJ, Turner APF (1989) Biosensors 4:299–311
Zhao JS, Wang Z, Wang M, Wang HS, He QP, Zhang H (2008) Talanta 74:1686–1691
Ertl P, Unterladstaetter U, Bayer K, Mikkelsen SR (2000) Anal Chem 72:4949–4956
Zhao JS, Wang Z, Fu CG, Wang M, He QP (2008) Electroanalysis 20:1587–1592
Kostesha N, Heiskanen A, Spegel C, Hahn-Hagerdal B, Gorwa-Grauslund M-F, Emneus J (2009) Bioelectrochemistry 76:180–188
Heiskanen A, Spégel C, Kostesha N, Lindahl S, Ruzgas T, Emnéus J (2009) Anal Biochem 384:11–19
Kostesha NV, Almeida JRM, Heiskanen AR, Gorwa-Grauslund MF, Hahn-Hagerdal B, Emneus J (2009) Anal Chem 81:9896–9901
Lovley DR (2008) Curr Opin Biotechnol 19:564–571
Kostesha N, Willquist K, Emneus J, van Niel EWJ (2011) Extremophiles 15:77–87
Ikeda T, Kurosaki T, Takayama K, Kano K (1996) Anal Chem 68:192–198
Baronian KHR, Downard AJ, Lowen RK, Pasco N (2002) Appl Microbiol Biotechnol 60:108–113
Spégel CF, Heiskanen AR, Kostesha N, Johanson TH, Gorwa-Grauslund MF, Koudelka-Hep M, Emneus J, Ruzgas T (2007) Anal Chem 79:8919–8926
Schröder U (2007) Phys Chem Chem Phys 9:2619–2629
Gregg BA, Heller A (1991) J Phys Chem 95:5970–5975
Vostiar I, Ferapontova EE, Gorton L (2004) Electrochem Commun 6:621–626
Timur S, Haghighi B, Tkac J, Pazarhoglu N, Telefoncu A, Gorton L (2007) Bioelectrochemistry 71:38–45
Alferov S, Coman V, Gustavsson T, Reshetilov A, von Wachenfeldt C, Hagerhall C, Gorton L (2009) Electrochim Acta 54:4979–4984
Coman V, Gustavsson T, Finkelsteinas A, von Wachenfeldt C, Hägerhäll C, Gorton L (2009) J Am Chem Soc 131:16171–16176
Hasan K, Patil SA, Górecki K, Leech D, Hägerhäll C, Gorton L (2012) Electrochemical communication between heterotrophically grown Rhodobacter capsulatus with electrodes mediated by an osmium redox polymer. Bioelectrochemistry. doi:10.1016/j.bioelechem.2012.05.004
Shkil H, Schulte A, Guschin DA, Schuhmann W (2011) Chemphyschem 12:806–813
Weibel DB, DiLuzio WR, Whitesides GM (2007) Nat Rev Microbiol 5:209–218
Lee JW, Kjeang E (2010) A perspective on microfluidic biofuel cells. Biomicrofluidics. doi:10.1063/1.3515523:041301
Qian F, Morse DE (2011) Trends Biotechnol 29:62–69
Wang H-Y, Bernarda A, Huang C-Y, Lee D-J, Chang J-S (2011) Biores Technol 102:235–243
Hou H, Li L, de Figueiredo P, Han A (2011) Biosens Bioelectron 26:2680–2684
Ertl P, Wagner M, Corton E, Mikkelsen SR (2003) Biosens Bioelectron 18:907–916
Gottschamel J, Richter L, Mak A, Jungreuthmayer C, Birnbaumer G, Milnera M, Brueckl H, Ertl P (2009) Anal Chem 81:8503–8512
MicroChemicals GmbH (2003) Processing image reversal resists. MicroChemicals GmbH, Ulm. http://www.microchemicals.eu/technical_information/image_reversal_resists.pdf. Accessed 16 Feb 2012
Fischer LM, Tenje M, Heiskanen AR, Masuda N, Castillo J, Bentien A, Emneus J, Jakobsen MH, Boisen A (2009) Microelectron Eng 86:1282–1285
Gao ZQ, Binyamin G, Kim HH, Barton SC, Zhang YC, Heller A (2002) Angew Chem Int Ed 41:810–813
Boles E, Zimmermann FK (1993) Curr Genet 23:187–191
Boles E, Zimmermann FK (1994) Mol Gen Genet 243:363–368
Heiskanen A, Yakovleva J, Spegel C, Taboryski R, Koudelka-Hep M, Emneus J, Ruzgas T (2004) Electrochem Commun 6:219–224
Skafte-Pedersen P, Sabourin D, Dufva M, Snakenborg D (2009) Lab Chip 9:3003–3006
Gao QA, Guo YY, Zhang WY, Qi HL, Zhang CX (2011) Sens Actuators B 153:219–225
Shin H, Kang C (2010) Bull Kor Chem Soc 31:3118–3122
Zór K, Vergani M, Heiskanen A, Landini E, Carminati M, Coman V, Vedarethinam I, Skafte-Pedersen P, Skolimowski M, Martinez Serrano A, Kokaia M, Ramos Moreno T, Ghio A, Svendsen WE, Dimaki M, Keresztes Z, Adamovski M, Wollenberger U, Sabourin D, Ferrari G, Raiteri R, Sampietro M, Dufva M, Emnéus J (2011) Real-time monitoring of cellular dynamics using a microfluidic cell culture system with integrated electrode array and potentiostat. In: Landers JP, Herr A, Juncker D, Pamme N, Bienvenue J (eds) Proceedings of the 15th International Conference on Miniaturized Systems for Chemistry and Life Sciences (μTAS2011), Seattle, Washington, USA, vol. 2, pp 1532–1535
Guschin DA, Castillo J, Dimcheva N, Schuhmann W (2010) Anal Bioanal Chem 398:1661–1673
Sabourin D, Skafte-Pedersen P, Søe MJ, Hemmingsen M, Alberti M, Coman V, Petersen J, Emnéus J, Kutter JP, Snakenborg D, Jørgensen F, Clausen C, Holmstrøm K, Dufva M (2012) JALA. doi:10.1177/2211068212461445
Acknowledgments
This work was supported by the EU FP-7 project EXCELL (NMP4-SL-2008-214706). Additional financial support is acknowledged by A. H. (Ørsted postdoctoral grant and Lundbeck Foundation grant no. R69-A6408), N. H. (EU FP-6 project MEST-CT-2004-514743), and L. G. (the Swedish Research Council project 2010-5031). We thank Prof. Eckhard Boles (Institute of Microbiology, Frankfurt, Germany) for sharing the ENY.WA and EBY44 strains with us.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Bioelectroanalysis with guest editors Nicolas Plumeré, Magdalena Gebala, and Wolfgang Schuhmann.
Arto Heiskanen and Vasile Coman contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 4.46 MB)
Rights and permissions
About this article
Cite this article
Heiskanen, A., Coman, V., Kostesha, N. et al. Bioelectrochemical probing of intracellular redox processes in living yeast cells—application of redox polymer wiring in a microfluidic environment. Anal Bioanal Chem 405, 3847–3858 (2013). https://doi.org/10.1007/s00216-013-6709-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-6709-4