
Abstract
We report the development of a hyphenated instrument with the capacity to quantitatively characterize aqueous suspended gold nanoparticles (AuNPs) based on a combination of gas-phase size separation, particle counting, and elemental analysis. A customized electrospray-differential mobility analyzer (ES-DMA) was used to achieve real-time upstream size discrimination. A condensation particle counter and inductively coupled plasma mass spectrometer (ICP-MS) were employed as downstream detectors, providing information on number density and elemental composition, respectively, of aerosolized AuNPs versus the upstream size selected by ES-DMA. A gas-exchange device was designed and optimized to improve the conversion of air flow (from the electrospray) to argon flow required to sustain the ICP-MS plasma, the key compatibility issue for instrumental hyphenation. Our work provides the proof of concept and a working prototype for utilizing this construct to successfully measure (1) number- and mass-based distributions; (2) elemental compositions of nanoparticles classified by size, where the size classification and elemental analysis are performed within a single experiment; (3) particle concentrations in both solution (before size discrimination) and aerosol (after size discrimination) phases; and (4) the number of atoms per nanoparticle or the nanoparticle density.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nanoparticles (NPs), especially in the colloidal form (i.e., dispersed in a fluid medium), are increasingly used in the production of consumer products, biomedical and diagnostic devices, drug delivery systems, and advanced materials for catalysis, coatings, and food packaging, among other uses [1–10]. For these applications, physical size and elemental composition are among the most important properties that influence the performance of nanobased products [11–15]. For instance, nanomedicines that use NPs as drug delivery vehicles are under development [11, 13, 14, 16–18]. Studies have shown that therapeutic performance is related to the size and chemical composition of NPs used in the formulations [12, 15, 19]. Traceable, quantitative analytical methods for determining size and chemical composition are necessary to accurately and precisely correlate these properties to efficacy and safety and are critical for the advancement of NP-based therapeutics. For particle size characterization, techniques based on physical measurements, especially classification prior to detection (e.g., chromatography, electrophoresis, and field flow fractionation), are widely used due to their capacity to enable size-resolved measurements [20–22]. To characterize elemental composition with high sensitivity, techniques such as mass spectrometry, optical emission spectrometry, X-ray photoelectron spectroscopy, and energy dispersive X-ray spectroscopy are used extensively [12, 23–28]. By combining the information obtained by multiple characterization methods, the material properties of NPs can be presented more completely and accurately.
In addition to applying complementary measurements independently [11, 16, 23, 25, 29–32], a recent advance is the development of real-time, quasi-simultaneous characterization methods through the hyphenation of complementary instrumentation: e.g., upstream size separation coupled with downstream elemental detection [20, 33, 34]. Hyphenation not only combines the advantages of using these instruments individually, but also offers a major advantage by providing real-time analysis of the material properties of NPs for a more synchronized data comparison. For example, a complex mixture of NPs can first be classified and subsequently analyzed, in order to differentiate among coexisting populations, with minimal perturbation to the native sample.
Differential mobility analysis (DMA), also called scanning ion mobility spectrometry, is a high-resolution size classification technique relevant to the analysis of discrete (dispersed) nanoscale species such as NPs, viruses, proteins, and DNA [35, 36]. For sampling from an aqueous solution, an aerosol generator, such as an electrospray (ES) device, can be combined with DMA, allowing the DMA size discrimination to be applied to particles as small as a few nanometers in diameter [11, 16, 30, 31, 35–40]. The major advantage of using electrospray-differential mobility analyzer (ES-DMA) is that the ES can generate well-controlled, monodisperse, sub-micrometer droplets containing a single particle per droplet [35–37, 41, 42]; thus, it is ideal for characterizing NPs at appropriate particle number concentrations (1010 to 1013/cm3) with resolution down to about 0.3 nm. In the classical application of ES-DMA, a condensation particle counter (CPC) is attached to detect aerosolized particles of a specific size, selected by controlling the applied voltage in the DMA classifier. In this configuration the technique is limited to providing aerodynamic size-resolved concentration, albeit with a very high degree of size resolution.
For quantitative elemental analysis, inductively coupled plasma mass spectrometry (ICP-MS) is the technique of choice, due to sensitivity at sub-nanograms per kilogram (parts per trillion) to micrograms per kilogram (parts per billion) levels [22, 43, 44]. By combining DMA with ICP-MS, it should be feasible to directly obtain size-resolved composition and mass-weighted size distributions of nanoscale species with high sensitivity. Previous studies have also shown the feasibility to measure the composition of aerosolized NPs directly by integrating ICP-MS with different types of aerosol generators (e.g., atomizer/nebulizer and laser ablation) [34, 43, 45]. Elemental composition, on a basis of single particle analysis, could be achievable for the NPs having sufficiently large size and at low number concentration in solution.[43, 46] We are aware of only two previously published studies in which ES-DMA has been directly hyphenated to ICP-MS: Kapellios et al. [33] and Carazzone et al.[39] partially addressed the problem of incompatibility of air with the ICP-MS plasma (air is typically used in ES and as the sheath flow in DMA). As we will show, gas incompatibility is the principal coupling issue for hyphenation of ES-DMA with ICP-MS.
The ES device requires air or other compatible conductive gases and cannot be operated using Ar due to the low electrical breakdown voltage of the latter. On the other hand, the ICP-MS operates with Ar, where air content must be minimized to sustain a stable plasma. Moreover, the high oxygen content present in air interferes with the detection and quantification of several key elements, such as S, so it is advantageous to minimize air entering the ICP. Hence, the air in the gas flow carrying the analyte must be exchanged with argon before introduction into the ICP-MS system. One partial solution [34] is to utilize the DMA itself as a gas converter (i.e., use a sufficient flow of Ar as the sheath gas in the DMA) to achieve sufficiently low air content to allow stable on-line ICP-MS measurements. Although these previous studies show it is possible to overcome the air/plasma stability issue by using the DMA as the sole gas converter [39], the necessity of using a higher sheath flow rate significantly restricts the operational size range of the DMA and decreases the concentration of aerosolized NPs delivered to the downstream ICP-MS detector. As a result, the gas-exchange efficiency is coupled to the size resolution and dynamic range of the DMA. Additionally, the residual air flow is still high enough to present an interference issue in the MS detection of specific elements.
Our primary objective is to develop and validate a high-resolution, hyphenated measurement system capable of providing quantitative information on the size-resolved elemental composition of engineered NPs and their functional coatings (where applicable). Through a systematic study of ES-DMA coupled with ICP-MS (denoted as ES-DMA/ICP-MS), we optimized the operational window (e.g., size range, sensitivity of elemental analysis, etc.) for this hyphenated system, and extended the measurement capability to obtain other important material properties (e.g., particle concentration and particle density). We designed and constructed a gas-exchange device (GED) installed just prior to the ICP, which affords the capacity to operate the DMA at the desired size range while achieving the gas-exchange efficiency necessary for high sensitivity elemental analysis. The GED concept is based on the membrane gas exchange apparatus described previously by Nishiguchi et al. [45] for direct monitoring of airborne particulates by ICP-MS. Gold nanoparticles (AuNPs) were chosen as the test material for the present studies, because of their widespread use in biomedical applications and because of our substantial prior work on this material [11, 16–18, 23, 29, 30, 47]. The GED was systematically studied to optimize efficiencies of gas exchange and particle transport. Additionally, the advantages of this technique are discussed for applications related to NPs and nanobased products.
Experimental
Materials
Commercially available AuNP suspensions (nominally 30 nm and 10 nm in diameter) were purchased from Ted Pella (Redding, CA).Footnote 1 A 20-nm polystyrene (PS) nanosphere size standard was purchased from Thermo Scientific (Madison, WI). Ammonium acetate (Sigma-Aldrich, St. Louis, MO) was used to adjust the ionic strength of samples for ES operation. Biological-grade (18 MΩ × cm) high-purity filtered deionized water (Aqua Solutions, Jasper, GA) was used for all sample preparation and dilutions.
Sample preparation
Citrate-stabilized AuNPs (1 mL) were centrifuged (MiniSpin Plus, Eppendorf North America, Hauppauge, NY) for one cycle (additional cycles prevented resuspension) under the following conditions: 30 nm AuNPs for 15 min at 9,660×g; 10 nm AuNPs for 45 min at 14,100×g. After centrifugation, the supernatant was removed, ammonium acetate solution (5 mmol L−1) was added for final Au concentrations of approximately 5–250 μg g−1, and samples were sonicated using a bath sonicator (model 1510, Branson, Danbury, CT) to aid resuspension of the particles.
Instrumentation
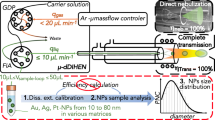
The hyphenated instrument (Fig. 1) comprises an ES-DMA for aerosol generation and size discrimination, a GED to exchange air for argon, a CPC to determine particle number concentration, and an ICP-MS for quantitative elemental analysis. Each component is described in more detail below.

Schematic diagram of the ES-DMA/ICP-MS hybrid instrument
Electrospray-differential mobility analyzer
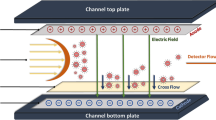
The ES (model 3480, TSI, Inc., Shoreview, MN) generates an aerosol of NPs by using a differential pressure to move a liquid dispersion through a fused silica capillary (40 μm inner diameter tip), after which the droplets are sprayed under an electric field into a stream of dry air. The aerosolized NPs acquire an equilibrium charge distribution due to a radioactive 210Po neutralizer. The NP aerosol stream (denoted as polydisperse flow) then enters the DMA (model 3085, TSI, Inc.), where the particles are classified based on electrical mobility under an applied electric field, with a sheath flow carrying NPs downstream. The trajectory of a charged particle within the DMA column is directly related to the particle mobility diameter, d p, m (Fig. 2A). As the voltage applied to the DMA is varied, particles of a specific size are allowed to exit the DMA [48, 49] and are counted by the CPC (model 3776, TSI, Inc.). The CPC uses butanol condensation to grow particles to an optically detectable size, and individual particles are counted by a photodetector. A Bertan power supply (205B-10R, Valhalla, NY) was used to apply voltage to the DMA. The electric field of the DMA and data acquisition for the CPC are controlled using a customized LabView program (National Instruments Corp., Austin, TX). The ES-DMA-CPC is used to obtain a particle number-based size distribution (i.e., particle number concentration in gas phase (N p) versus selected d p, m). The step size used in the particle size measurement is 0.2 nm, and the time interval between each step size is 10 s. Sheath flow rate (Q sh = 10 L min−1 in our experiments) and sample flow rate (Q samp) of the DMA are controlled by a mass flow controller (MFC, MKS Instruments, Andover, MA) and a customized laminar flow element (CME, Davenport, IA), respectively. The resolution of the particle size measurement is 0.3 nm. [25, 32, 50]

(A) Schematic representation of size discrimination process by ES-DMA. (B) Schematic diagram of the GED, where PGM is the porous glass membrane
Gas-exchange device
Table 1 shows the instrumental settings for the GED. A porous Al2O3⋅SiO2 glass membrane (PGM) with 100 nm diameter pores (SPG Technology Co., Ltd., Japan) is housed in a custom-made Pyrex glass tube (Fig. 2B). The GED inlet (sample flow) and outlet flow are in line with the membrane (inner tube), while separate Ar inlet (sweep flow) and outlet ports are perpendicular to the flow through the membrane. Because of the concentration gradient of gases between inner and outer tubes of the GED, air enters the GED inlet, diffuses through the pores of the membrane, and exits with the sweep gas flow; simultaneously, Ar diffuses into the membrane. Thus, air is replaced by Ar via diffusion, and the flow at the GED outlet is predominantly Ar. The MFC was used to control the Ar sweep flow into the GED.
The percent O2 at the GED outlet (C o, 2) was measured using a paramagnetic analyzer (OXYMAT 6, Siemens, San Luis Obispo, CA). The percent O2 was used to calculate the exchange efficiency, EffGED (percent air replaced by Ar) relative to the initial measurement of the percent O2 in the sample flow just before entering the GED, C o, 1 (no Ar sweep flow).
The Q samp was varied from 0.25 to 1 L min−1, and the Ar sweep flow (Q sweep)was varied from 0 to 6 L min−1. The flow rate at the GED outlet was measured by a DryCal flow meter (ML-800-24, Bios International Corp., Butler, NJ) set for Ar. The relative standard uncertainty (precision) of the O2 measurements is typically less than or equal to 0.03 %.
Inductively coupled plasma mass spectrometer
An ICP-MS (7700x, Agilent Technologies, Santa Clara, CA) was used to monitor 197Au (Table 2). The ES-DMA was coupled to the ICP-MS via the GED. Specifically, the output of the DMA was connected to the GED inlet with Tygon tubing (I.D., 0.64 cm). With the spray chamber of the ICP-MS removed, the outlet of the GED was connected with Tygon tubing (ID, 0.95 cm) to the ball joint at the end of the torch via a 12 × 5 Pyrex glass socket joint. Other parameters not listed in Tables 1 and 2 are included in Table 3.
Hybrid instrument operation (ES-DMA/ICP-MS)
For sample analysis using the hyphenated instrument, the voltage applied to the DMA was increased in stepping mode, and the particle mobility diameter (i.e., determined by equating electrostatic force to Stokes drag)[25, 49] was measured from d p, m = 2 to 40 nm. Ar was used as the sheath gas in DMA to dilute the extent of air present in the sample flow without reducing the NP concentration. Initially, the particles exiting the DMA were directed to the CPC (denoted as ES-DMA-CPC or classical mode) to determine the number-based particle size distribution (PSD—i.e., N p versus selected d p, m). Subsequently, the particle stream was delivered to the ICP-MS, and acquisition mode was set to time-resolved acquisition (TRA) to monitor 197Au signal intensity as the DMA voltage was increased (i.e., 197Au counts versus the scanning time (t s)), where the d p, m, or DMA voltage, can be correlated as a step function of t s (Eq. (2)), allowing particles of a specific diameter to exit the DMA at the selected voltage and to be atomized by the plasma and detected by the ICP-MS. The ES-DMA/ICP-MS mode was used to obtain a mass-weighted PSD. The step size used in the particle size measurement was 0.2 nm, and the time interval between each step size was 20 s. A relative standard uncertainty of 7 % is estimated for mass distribution measurements obtained in coupled mode; this value is based on the mean standard deviation of 40 measurements performed at each size step. Note that the brackets on the right side of Eq. (2) indicate the floor function, which rounds the quotient down to the nearest whole number.
Results and discussion
Study of GED
Gas-exchange efficiency
As shown in Fig. 3A, decreasing Q samp, or increasing Q sweep, increases EffGED significantly. When Q sweep > 3 L min−1, EffGED reaches 98, 85, and 57 % through exchange at Q samp of 0.25, 0.5, and 1.0 L min−1, respectively. The results of gas-exchange efficiency (Fig. 3A) can be interpreted mechanistically by considering both the free diffusion of gas molecules and their penetration through the pores of the GED membrane. Since the pore size of the membrane (100 nm) is at least 100× larger than the size of the gas molecules (principally Ar, N2, and O2 in this system), the gas penetration resistance due to the steric resistance of membrane pores is negligible. Hence, the mechanism of gas exchange is dominated by molecular diffusion. We investigated the effect of Q samp and Q sweep on the diffusion flux of gas molecules by applying Fick’s law of diffusion [49]:

Analysis of the gas-exchange device (GED). (A) EffGED versus Q sample and Q sweep. (B) Dimensional analysis of EffGED versus gas-exchange factor (Ω). (C) Efficiency of particle transport through the GED (N p, GED/N p, samp) as a function of Q sweep and nominal particle size. The native AuNP concentration was ≈250 μg g–1
Here, J Ar and J air are the diffusion rates of Ar and air through the membrane, respectively, and t mem is the retention time of sample flow through the PGM. According to Eq. (3), J Ar, J air, and t mem are proportional to EffGED. Increasing Q sweep should increase the concentration gradient between the sweeping flow and the sample flow, resulting in an increase in both J Ar and J air. Decreasing Q samp increases t mem (i.e., allowing more time for gas diffusion). Hence, decreasing Q samp, or increasing Q sweep, will increase EffGED. Our experimental results (Fig. 3A) support this mechanism.
We define a gas-exchange factor (Ω) to quantify the effect of gas diffusion on EffGED. As shown in Eq. (4) (a detailed derivation is provided in the Electronic supplementary material (ESM)), Ω is proportional to the diffusion flux and can be summarized as a function of the length (L mem) and the radius (r mem) of the PGM, respectively (in units of cm), Q samp, and Q sweep.
As shown in Fig. 3B, increasing Ω pushes EffGED toward 100 %, suggesting a longer retention time, or a higher concentration gradient, is beneficial for achieving higher Eff ,GED. The result also indicates that L mem and r mem could be increased to maintain higher Ω, if an increased value of Q samp is required for measurement resolution or sensitivity.
Particle transport
In addition to investigating EffGED, we also evaluated the potential influence of Q samp and Q sweep on the loss of particles during transport. However, even without using the GED, the particle concentration from the ES process was reduced significantly by reducing Q samp from 1 to 0.25 L min−1. We attribute this loss to the deposition of highly charged AuNPs generated by the ES onto the wall of the orifice plate, thus failing to reach the neutralization chamber. This effect is apparent at low carrier flow rates. ICP-MS requires Q samp of ≈1 L min−1 to ensure sample penetration through the plasma, thus Q samp at the GED inlet must be at least 1 L min−1 for this application. We therefore set Q samp equal to 1 L min−1 for the measurement of AuNP transport.
Particle loss due to diffusion and/or deposition onto the surface of the GED was quantified based on the measured particle counts in sample flow using the ES-DMA-CPC configuration (Fig. 3C). Number concentration of size-classified AuNPs was measured under various Q sweep (0 to 6 L min−1) at a Q samp of 1 L min−1. Particle loss through the GED, P L [(1 − (N p, GED/N p, samp), where N p, samp and N p, GED are N p measured before and after the GED, respectively], was determined using AuNPs of two nominal diameters: 30 and 10 nm (denoted as 30AuNP and 10AuNP, respectively). Unlike gas molecules, the size of the particles used in our experiment are within one order of magnitude of the pore size of the PGM. The steric hindrance of the pores may therefore restrict NP diffusion toward the outer chamber of the GED—a beneficial effect of size discrimination (i.e., gas molecules can diffuse through the membrane, but generally not NPs).
For 30AuNPs, particle loss due to the GED was negligible (≈1.5 % when Q sweep = 0 L min−1). Interestingly, Ar sweep flow through the GED increased the particle number concentration beyond the baseline value (measured without the GED) by ≈4 % at Q sweep = 3 L min−1 and 8 % at Q sweep = 6 L min−1. The reason for the increase in particle concentration is not entirely clear. However, it could be due to a slight increase in the Ar content of the carrier flow post-GED (i.e., less diffusion loss due to the denser Ar carrier gas), or possibly due to the uncertainty in the measurement of particle concentration (typically ≈3 % but can be as high as 20 % under some conditions) [31, 35]. Details of the calculation of particle loss are described in Section 2 of the ESM.
For 10AuNPs, particle loss due to the GED was >23 %. This relatively high loss could be the result of increased diffusional deposition of the smaller particles and/or an increase in penetration through the pores of the GED membrane itself. Based on calculations of diffusion loss [49], diffusion accounts for ≈4 % of the particle loss for 10AuNPs and 1 % for 30AuNPs (details in ESM). Thus, particle loss for the 30AuNPs is attributed principally to diffusion, but the most of the particle loss for the 10AuNPs can be attributed to penetration through the membrane pores. We also observed that increasing Q sweep increased the particle number concentration relative to the baseline by ≈4 % at Q sweep = 3 L min−1 and >7 % at Q sweep = 6 L min−1, similar to the increase for the 30AuNPs at the same value of Q sweep. Hence, increasing Q sweep (or the relative pressure outside the membrane) may suppress the diffusion loss of particles (through the membrane or in transport).
We have demonstrated the proof of concept for using the GED to discriminate the diffusion of gas molecules from AuNPs, and then to achieve sufficient gas-exchange efficiency without substantial loss of particles during transport. Based on the particle size involved, and the tolerance of ICP-MS to air (i.e., an air content below 10 % is necessary for the stability of the plasma), parameters such as the length and pore size of the membrane can also be adjusted for optimization. This is the key advantage of using a modular GED instead of relying on the DMA alone for reducing air content. For example, in order to avoid exceeding the threshold of electrical breakdown for the Ar sheath gas inside the electrostatic classifier, the DMA voltage has to be operated under ≈−3 kV (based on our experimental setup), which is not applicable for NPs having d p, m > 40 nm. With the assistance of the GED, we were able to perform measurements for d p, m > 40 nm while maintaining stable plasma operation (i.e., operation at lower Q sh while maintaining the air content below 10 % for the ICP). In the next section, we discuss the application of the GED to the hyphenation of ES-DMA and ICP-MS.
Hybrid instrument (ES-DMA-GED/ICP-MS)
Particle size distribution with hyphenated elemental analysis
In this section, we discuss characterization of AuNPs using our customized ES-DMA system coupled to the ICP-MS through the GED. The ES-DMA was operated at an aerosol flow of 1 L min−1 to ensure that particles from the ES were successfully delivered to the DMA. We chose Q sh = 10 L min−1 to ensure sufficiently low air content delivery to the ICP. Q sweep of 3 L min−1 was used for the hyphenated measurements. Although the ICP-MS can operate at Q sh = 10 L min−1 without using the GED, the addition of the GED can reduce the level of air to improve the plasma stability and potentially reduce interference from O species.
As shown in Fig. 4A, the PSD of the 30AuNP sample (250 μg g−1) was measured by increasing the voltage applied to the DMA in stepping mode. Figure 4A shows the TRA of the particle number concentration and mass signal intensity. The inset shows the number-based PSD obtained using the CPC (red line) and the mass-based PSD obtained using the ICP-MS (blue line). Two peaks were observed using the CPC detector: the first peak at 9.4 nm is attributed to salt residue observed in our previous studies [16, 25, 32], and the second peak at 31.8 nm is assigned to the AuNPs. Using a salt correction model described previously [25], the electrical mobility diameter of 30AuNPs was determined to be 31.5 nm.

30AuNPs measured by ES-DMA sample introduction into the ICP-MS and comparison with the result using CPC. (A) TRA mode. (B) Particle size distribution. The particle number concentration in the gas phase (N p) was acquired at each size step (10 s for each 0.2 nm) using the ES-DMA-CPC (red dashed line). Subsequently, the particle flow was redirected into the ICP-MS and the 197Au signal intensity was acquired at each time step (each 0.2 nm increase for 20 s) using the ES-DMA/ICP-MS (blue solid line). Concentration of 30AuNPs was ≈250 μg g−1 (concentration factor = 5)
By combining the data obtained from the CPC and the ICP-MS, the elemental composition of particles can be identified in a size-dependent manner. After the CPC data had been acquired, the direction of particle flow was switched from the CPC toward the ICP-MS, and the 197Au signal intensity was acquired at each size step using the ES-DMA/ICP-MS with the ICP-MS set to TRA mode. In Fig. 4A, AuNPs are clearly distinguishable from the salt residue, based on the coincidence of the peak (enlarged in Fig. 4B) at ≈32 nm (CPC) and the 197Au peak (ICP-MS). However, there is no 197Au signal corresponding to the peak at 9.4 nm, confirming the peak consists only of salt residue (mainly sodium ≈20 ppm and potassium ≈30 ppm found in the 30AuNPs using the semi-quantitative mode of the ICP-MS). Thus, this hybrid instrument can distinguish the components in a mixed sample based on elemental composition of size-separated particles.
The hyphenated method described here provides a promising way to resolve the issue of salt residue that frequently occurs during ES-DMA analysis [25]. It is well known that salt residue from the ES process interferes with ES-DMA measurements; therefore, it is necessary to remove non-volatile salts and add volatile ammonium acetate solution prior to ES-DMA measurements. If the ES-synthesized (artificial) salt particles have a size that is close to, or even larger than, the size of the NP analyte, the number-based PSD of the NPs will be obscured by the salt peak [25]. The ICP-MS detector, on the other hand, can successfully distinguish the population of artificial salt particles from the AuNPs based on elemental analysis. Hence, we can still measure an accurate PSD of NPs while avoiding the deleterious effects of non-volatile salt removal (e.g., centrifugation-induced aggregation). Also, the capacity to obtain mass-based PSD for comparison to the number-based distribution provides the opportunity for a more sophisticated analysis.
Measurement of the concentration of AuNPs in solution
This hyphenation system can also be used as a complementary technique for measuring the concentration of particles in solution, N p, l, a key factor related to the dose of NP-based therapeutics. Since the particles we measured represent a small fraction of the population sampled by ES-DMA, N p, l can be calculated by considering the charge distribution, transfer function, liquid flow rate, and other parameters. We used 30AuNP suspensions at various concentration factors (N p, l/N p, l, 0, where N p, l, 0 is the as-received N p, l) for calibration. As shown in Fig. 5A, we obtain linear response for both detectors: R 2 = 0.9897 for the CPC and R 2 = 0.9984 for ICP-MS. Because of the linear correlations, we were able to determine the absolute number (by CPC) and mass (by ICP-MS) concentrations of AuNPs in solution, with appropriate calibration standard materials: PS standard colloid and NIST 8012 AuNP reference material, respectively. PS standard colloid, with a known size and population, was used to perform an external calibration of absolute number of particles measured by ES-DMA-CPC. The N p, l of AuNPs in solution can be calculated using Eq. (5):
where N p, g is the gas-phase particle concentration before size discrimination, which can be calculated by integrating the full particle size distribution, after considering charge correction and transfer function.[30, 37] N p, g, PS and N p, l, PS are the N p, g and N p, l of PS, respectively (details of the calculation of N p, g, N p, g, PS, and N p, l, PS are provided in the ESM). Complementarily, the total mass of 197Au in solution can also be obtained by calibration with AuNP standard reference materials [51] (see Section 4 of the ESM). By measuring the as-received sample (i.e., concentration factor = 1), we calculated the number and mass concentrations of AuNPs in solution and obtained values of ≈2.0 × 1011 cm−3 and ≈48 ppm, respectively.

Quantitative analysis. (A) Calibration of AuNP suspensions (using 30AuNPs) as a function of the concentration factor using ES-DMA sample introduction into the CPC or into the ICP-MS. (B) Calculation of the average density of a AuNP (ρ AuNP) and number of Au atoms per particle (N Au), based on the results of (A)
Measurement of the density of AuNPs
Next we calculate the number of Au atoms per particle and the density of a single “average” particle from the results of Fig. 4B. The number of 197Au atoms per particle, N Au, can be obtained by dividing the total 197Au atoms present in solution (N Au, total) by N p, l, s. The average density of a AuNP, ρ AuNP, can then be calculated using Eq. (6):
where V AuNP is the volume of a single AuNP. Assuming that the AuNP is spherical (a reasonable approximation in this case [51]), Fig. 5B shows the results of ρ AuNP for various concentration factors. Excluding the first outlier (at concentration factor = 0.2), the mean value for ρ AuNP is 19.6 ± 2.0 g cm−3, which is in good agreement with the literature value for bulk Au (19.3 g cm−3). The corresponding N Au is ≈(9.8 ± 1.0) × 105. Reported uncertainties represent the standard deviation of four measurements at different concentration factors.
The methodology proposed here would be very useful for supporting other types of instrumentation requiring an accurate value of particle density, such as disc centrifugation and analytical centrifugation. For these centrifugation-based measurement methods, the principle to differentiate particle size is based on the mass of the NPs. However, the density of NPs is usually unknown and always assumed to be equal to the value of the corresponding bulk material. Although our results show that AuNP density is close to the value of bulk Au, this might not be true for other types of NPs, due, for instance, to differences in synthetic processes. Take amorphous silica NPs for example, the density can vary from 1.8 to 2.3 g cm−3 [52] and is generally less than the bulk value (≈2.65 g cm−3).
Applications
The hybrid instrument developed in this study is applicable to a broad range of NP-based materials, including environmental, biological, medicinal, and energy-related materials. There is an interest in analytical characterization of metal-containing NP colloids, both engineered and naturally occurring, for environmental studies of particle transport, trace metal transport via NPs, and water quality effects [53, 54], in which accurate and precise characterization of the size of particles and their aggregates, as well as their chemical composition, is critical.
This hyphenated technique has limitations with respect to the size range, concentration, and elements that can be detected. For example, purely organic materials cannot be quantified using ICP-MS. Depending on the DMA column used, the ES-DMA can only classify particles in the size range from ≈3 to ≈1,000 nm, but this covers most of the nanoscale regime and many particles of technological interest. In this study, the particle number concentration could be calibrated up to a level equivalent to Au mass fraction of ≈10 μg g−1. Because the detection limit of the batch ICP-MS is much lower, with ES-DMA upstream classification, we can obtain the elemental composition of “size-defined” NPs (e.g., ±0.3 nm in diameter for 30AuNP). This will be especially useful for analyzing the elemental composition of NP samples having different mobility sizes (e.g., 10AuNPs mixed with 30 nm SiO2 NPs) or the AuNPs containing impurities that are size dependent.
Conclusions
We have developed a prototype hyphenated instrument to characterize liquid-dispersed NPs based on upstream size separation coupled with quantitative elemental analysis and particle counting. A modular GED can improve conversion of air flow from the ES to Ar flow for ICP-MS analysis and also expand the operational size range for the hyphenated technique. By combining the data obtained from the CPC and the ICP-MS using ES-DMA sample introduction, the hybrid instrument can provide real-time results on particle size distributions with quantitative elemental composition, population of NPs in solution, the number of atoms per particle, and the average density of individual NPs. This is a powerful measurement technique for the simultaneous characterization of particle size and elemental composition, with relevance for a wide range of nanotechnology applications.
Current work is focused on optimization of methodology for analysis of more complex NP systems, such as core/shell particles and particle–ligand conjugates, thus expanding the technique to a broader range of real-world applications. Specific issues requiring further research include reduction of the air (particularly O2) content, which interferes with the determination of S and other key elements, and validation of calibration methods for a broader range of nanomaterials.
Notes
The identification of any commercial product or trade name does not imply endorsement or recommendation by the National Institute of Standards and Technology.
References
Henig RM (2007) Our silver-coated future. OnEarth. Fall 2007
Jin R (2012) Nanotechnology Review 1:31–56
Fakruddin M, Hossain Z, Afroz H (2012) Journal of Nanobiotechnology 10:31
Conde J, Rosa J, Lima JC, Baptista PV (2012) Int J Photoenergy. doi:10.1155/2012/619530
Tiwari PM, Vig K, Dennis VA, Singh SR (2011) Nanomaterials 1:31–63
Riggio C, Pagni E, Raffa V, Cuschieri A (2011) J Nanomaterials. doi:10.1155/2011/164506
Duncan TV (2011) J Colloid Interf Sci 363:1–24
Somasundaran P, Mehta SC, Rhein L, Chakraborty S (2007) MRS Bull 32:779–786
Sharrna P, Brown S, Walter G, Santra S, Moudgil B (2006) Adv Colloid Interfac 123:471–485
Zhang S, Sun D, Fu YQ, Du HJ (2003) Surf Coat Tech 167:113–119
Tsai DH, Elzey S, DelRio FW, Keene AM, Tyner KM, Clogston JD, MacCuspie RI, Guha S, Zachariah MR, Hackley VA (2012) Nanoscale 4:3208–3217
Aravind A, Veeranarayanan S, Poulose A, Nair R, Nagaoka Y, Yoshida Y, Maekawa T, Kumar D (2012) BioNanoScience 2:1–8
McNeil SE (2011) Characterization of nanoparticles intended for drug delivery. Springer, New York
Dobrovolskaia MA, Mcneil SE (2007) Nat Nanotechnol 2:469–478
Jokerst JV, Lobovkina T, Zare RN, Gambhir SS (2011) Nanomedicine-Uk 6:715–728
Tsai DH, DelRio FW, Keene AM, Tyner KM, MacCuspie RI, Cho TJ, Zachariah MR, Hackley VA (2011) Langmuir 27:2464–2477
Paciotti GF, Kingston DGI, Tamarkin L (2006) Drug Develop Res 67:47–54
Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L (2004) Drug Deliv 11:169–183
Baptista P, Pereira E, Eaton P, Doria G, Miranda A, Gomes I, Quaresma P, Franco R (2008) Anal Bioanal Chem 391:943–950
Bolea E, Jiménez-Lamana J, Laborda F, Castillo J (2011) Anal Bioanal Chem 401:2723–2732
Gautier E, Roberti M, Gettar R, Jiménez Rebagliati R, Batistoni D (2007) Anal Bioanal Chem 388:499–503
Helfrich A, Bettmer J (2011) Int J Mass Spectrom 307:92–98
Elzey S, Tsai D-H, Rabb S, Yu L, Winchester M, Hackley V (2012) Anal Bioanal Chem 403:145–149
Tsai DH, Huang TJ (2002) Appl Catal Gen 223:1–9
Tsai DH, Zangmeister RA, Pease Iii LF, Tarlov MJ, Zachariah MR (2008) Langmuir 24:8483–8490
Cho TJ, Zangmeister RA, MacCuspie RI, Patri AK, Hackley VA (2011) Chem Mater 23:2665–2676
Scheffer A, Engelhard C, Sperling M, Buscher W (2008) Anal Bioanal Chem 390:249–252
Sanchez SI, Small MW, Zuo J-m, Nuzzo RG (2009) J Am Chem Soc 131:8683–8689
Tsai D-H, Davila-Morris M, DelRio FW, Guha S, Zachariah MR, Hackley VA (2011) Langmuir 27:9302–9313
Tsai DH, Cho TJ, DelRio FW, Taurozzi J, Zachariah MR, Hackley VA (2011) J Am Chem Soc 133:8884–8887
Pease LF, Tsai DH, Brorson KA, Guha S, Zachariah MR, Tarlov MJ (2012) Anal Chem 83(5):1753–1759
Tsai D-H, DelRio FW, MacCuspie RI, Cho TJ, Zachariah MR, Hackley VA (2010) Langmuir 26:10325–10333
Kapellios EA, Pergantis SA (2012) J Anal Atom Spectrom 27:21–24
Myojo T, Takaya M, Ono-Ogasawara M (2002) Aerosol Sci Tech 36:76–83
Pease LF (2012) Trends Biotechnol 30:216–224
Guha S, Li M, Tarlov MJ, Zachariah MR (2012) Trends Biotechnol 30:291–300
Tsai DH, Pease LF 3rd, Zangmeister RA, Tarlov MJ, Zachariah MR (2009) Langmuir 25:140–146
Pease LF 3rd, Sorci M, Guha S, Tsai DH, Zachariah MR, Tarlov MJ, Belfort G (2010) Biophys J 99:3979–3985
Carazzone C, Raml R, Pergantis SA (2008) Anal Chem 80:5812–5818
Hogan CJ, Kettleson EM, Ramaswami B, Chen DR, Biswas P (2006) Anal Chem 78:844–852
Li MD, Guha S, Zangmeister R, Tarlov MJ, Zachariah MR (2011) Aerosol Sci Tech 45:849–860
Li M, Guha S, Zangmeister R, Tarlov MJ, Zachariah MR (2011) Langmuir 27:14732–14739
Gschwind S, Flamigni L, Koch J, Borovinskaya O, Groh S, Niemax K, Gunther D (2011) J Anal Atom Spectrom 26:1166–1174
Kovacs R, Nishiguchi K, Utani K, Gunther D (2010) J Anal Atom Spectrom 25:142–147
Nishiguchi K, Utani K, Fujimori E (2008) J Anal Atom Spectrom 23:1125–1129
Suzuki Y, Sato H, Hikida S, Nishiguchi K, Furuta N (2010) J Anal Atom Spectrom 25:947–949
Dobrovolskaia MA, Patri AK, Zheng J, Clogston JD, Ayub N, Aggarwal P, Neun BW, Hall JB, McNeil SE (2009) Nanomedicine: Nanotechnology, Biology, and Medicine 5:106–117
Kim SH, Mulholland GW, Zachariah MR (2007) J Aerosol Sci 38:823–842
Hinds WC (1999) Aerosol technology: properties, behavior, and measurement of airborne particles, 2nd edn. Wiley, New York
Mulholland GW, Donnelly MK, Hagwood CR, Kukuck SR, Hackley VA, Pui DYH (2006) J Res Natl Inst Stan 111:257–312
National Institute of Standards and Technology Report of Investigation RM8011–RM8013 2008
Bell NC, Minelli C, Tompkins J, Stevens MM, Shard AG (2012) Langmuir 28:10860–10872
Alvarez PJJ, Colvin V, Lead J, Stone V (2009) ACS Nano 3:1616–1619
Hassellöv M, Lyvén B, Haraldsson C, Sirinawin W (1999) Anal Chem 71:3497–3502
Acknowledgments
This research was performed while S.E. held a National Research Council Research Associateship Award at NIST. The authors thank Yonggao Yan and Mindong Li at NIST for their help with developing the customized DMA program. The authors thank Robert Cook and Julien Gigault at NIST, and Prof. Michael Zachariah at the University of Maryland, for manuscript review and helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 569 kb)
Rights and permissions
About this article
Cite this article
Elzey, S., Tsai, DH., Yu, L.L. et al. Real-time size discrimination and elemental analysis of gold nanoparticles using ES-DMA coupled to ICP-MS. Anal Bioanal Chem 405, 2279–2288 (2013). https://doi.org/10.1007/s00216-012-6617-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6617-z