
Abstract
The methodology for separations of saccharides in standard electrophoretic systems has been transferred to the short-capillary electrophoresis format. The laboratory-designed apparatus used employs a quartz capillary with an internal diameter of 10 μm, a total length of 10 cm, and an effective length of 4 cm, in combination with contactless conductivity detection. It has been applied to separations of neutral mono- and disaccharides. The saccharides are separated in the anionic form, in solutions of alkali hydroxides, namely, KOH, NaOH, and LiOH. The separation of a model mixture of five saccharides (sucrose, lactose, glucose, fructose, and ribose) takes less than 1 min, the LOD equaling 15, 35, 19, 17, and 24 mg L−1 and the LOQ equaling 52, 117, 63, 53, and 79 mg L−1 for sucrose, lactose, glucose, fructose, and ribose, respectively. The technique developed has been used to determine sucrose, glucose and fructose in high-energy drinks. The separation is finished within less than 50 s; the saccharide contents determined are identical with the declared values within the reliability interval in most cases, the RSD value being mostly less than 2 %. In general, the separation system developed is very convenient for rapid analyses of large sets of similar samples, e.g., in product quality control or environmental monitoring.

Rapid determination of saccharides by CZE
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Analytical practice requires many routine analyses of large sets of samples, e.g., in the production control, in monitoring various environmental components, in clinical analysis, etc. These analyses must be sufficiently reliable but also simple, rapid, and inexpensive. The instrument developed and described in the present paper meets these requirements, and its functioning is described in detail on the monitoring of selected saccharides in commercial drinks.
The kinds of simple saccharides and their amounts contained in drinks and foodstuffs are important for checking of the foodstuff energy values, the control of their production procedures, the time of their storage, and possibly also for disclosing illegal approaches to these fields [1]. The structures of mono- and disaccharides are very similar and thus complex analyses of saccharide mixtures require the use of high-performance separation techniques.
For non-derivatized saccharrides, HPLC with refractive index detection (RID) [2–4] is used. Unfortunately, the RID suffers from a lack of sensitivity, selectivity, and is not robust enough. It also requires a long time for the baseline stabilization after a change in the experimental conditions [5]. An alternative method is high-pressure liquid chromatography (HPLC) with pulsed amperometric detection (PAD) [6–8]; the PAD is a sufficiently sensitive method, however, its use suffers from a typical problem of amperometric detection—gradual deterioration of the activity of the detection electrode, which leads to a poorer detection reproducibility. Furthermore, HPLC separations are rather slow (usually not shorter than ca. 10 min) and the sample pretreatment is demanding.
Much easier solutions to the problem of analyses of simple neutral saccharides are offered by the techniques of capillary electrophoresis (CE) [9–13]. Neutral mono- and disaccharides can be separated in strongly alkaline separation electrolytes with pH values higher than 12. The semi-acetal groups dissociate in these electrolytes and thus the saccharides are separated in the anionic form. The main advantages of the CE analysis involve a simple preparation of the samples, high separation efficiency, and a short separation time. Furthermore, the separation time can further be substantially decreased by using a short separation pathway [14]. It follows from the relationship for the migration time (t m),
where L is the capillary length, u eff is the analyte effective electrophoretic mobility, u eof is the electro-osmotic flow mobility, and U el is the separation voltage, that a shortening of the separation pathway to one half leads to the shortening of the analyte migration time to one quarter, at a constant separation voltage.
The design of commercial electrophoretic instrumentation does not permit the use of very short capillaries; e.g., the often employed Agilent instruments require capillaries at least 30 cm long [15]. To make separations faster, the sample can be introduced into the outlet end of the capillary; this technique is called the short-end injection. Another possibility is the use of a special laboratory device developed for electrophoretic separations in short capillaries [14]. The present paper describes one of such devices and demonstrates its advantages on the example of determination of saccharides in commonly available high-energy drinks.
Experimental
A principal experimental problem of separations in short capillaries lies in limited possibilities of moving the capillary. The capillary cannot be manipulated in the same way as capillaries several tens of cm long which are used in standard electrophoretic instruments. Therefore, the apparatus used in the present work has been designed in such a way that all the experimental steps needed, primarily the sample introduction and the rinsing of the capillary, can be carried out without any moving of the capillary. The apparatus has been assembled using mostly standard, commercially available components; for a scheme see Fig. 1.

A scheme of the apparatus for electrophoretic separations in a short capillary. 1, Separation capillary (Polymicro Technologies, USA); 2, sample introduction vessel, modificated Tee connector No. 54989 (Supelco, USA) with a grounding electrode; 3, sample outlet vessel, 5 mL sample vessel, cat. No. K000737 (P-Lab., Czech Republic) with a high-voltage electrode; 4, detection cell; 5, PTFE tubing, 1.58 and 0.8 mm in the outer and the internal diameter, respectively (Supelco, USA); 6, six-way sample introduction valve C102M (Labio, Czech Republic); 7, filling of the sample introduction loop; 8, piezoelectric sample introduction pump, type mp6, operated by an mp-x controller (Bartels Mikrotechnik, Germany); 9, membrane rinsing pump N86KN.18 (Laborport, USA)
Electrophoretic part of the apparatus
The separation capillary (1) is firmly fixed by standard screws and silicone rubber seals between the sample introduction “T”-shaped vessel (2) and the plastic outlet vessel (3). The detection cell (4) is placed close to the outlet vessel and the capillary is pressed inside it to the two electrodes of the contactless conductivity cell. These components are placed on the carrier plastic plate (denoted by dashed line in Fig. 1). The detection cell and the outlet vessel can be moved horizontally in the grooves cut in the carrier plate. This permits the use of capillaries of various lengths and variations in the capillary effective length, i.e., in the distance between the sample introduction end and the detection cell.
The measurements have been performed using a standard quartz capillary, 10 μm in internal diameter, total and effective length of 10 and 4 cm, respectively, if not stated otherwise. The total length of a capillary with this internal diameter should not be shortened, because large currents would pass through highly conductive hydroxide solutions and undesirable Joule heat would be generated. A capillary with a smaller internal diameter, e.g., 5 μm (see, e.g., [12]), would permit its shortening, however, the danger of its blocking would increase as would the demands on the detection sensitivity.
The sample introduction into the capillary is based on the principle described in ref. [16]. The sample introduction end of the capillary is inserted to a distance of ca. 0.5 mm into a standard PTFE tubing (5) in the sample introduction vessel. The PTFE tubing is connected with the outlet of the electronically operated, six-way sample introduction valve (6) equipped with the sampling loop with a volume of 15 μL. The sampling loop is filled with a syringe (7).
The sample is introduced into the capillary by simultaneous setting of the sampling valve in the position “INJECT” and switching on the piezoelectric micro-pump (8). This pump passes the separation electrolyte from the supply vessel (not shown) into the sampling part of the apparatus. The defined sample volume is carried by the stream of the separation electrolyte along the sampling end of the capillary. The time of the sampling is thus determined by the time for which the sample solution zone passes along the capillary; the separation electrolyte flows into the waste during the sample introduction. The sampling parameters, i.e., the time of activation of the sampling pump and the flow rate, have been adjusted to attain a time of contact of the sample zone with the capillary sampling end for ca. 0.3 s (as measured by injecting a small air bubble into the sample solution—consequently, the sample dispersion is not considered) and to transfer the sample zone sufficiently far from the capillary sampling end. The capillary is rinsed with the separation electrolyte between individual analyses, by decreasing the pressure in the outlet vessel using the membrane pump (9).
Platinum wires serve as the electrophoretic electrodes, the grounding electrode being placed in the sampling vessel and the high-voltage one in the outlet vessel. The separations have been carried out at a voltage of 5 kV, supplied by a high-voltage source, PS 350/5000 (Stanford Research Systems, USA); the separation field strength in the capillary, 500 V cm−1, is common in CE separations.
Electronic part of the apparatus
The contactless conductivity detector (C4D) contains the detection cell (4) laboratory-made according to ref. [17], consisting of two aluminum foil semitubular electrodes 1 mm wide, with a 1 mm gap between them. The electrodes are cemented in a groove whose width equals the outer diameter of the separation capillary and is cut in a plexiglass platelet. The capillary is pressed onto the electrodes by another plexiglass platelet. The cell is shielded from the surroundings by a metallic box. The detector electronic circuitry is based on the circuit described in refs. [18,19]. The input sine-wave a.c. signal has a frequency of 320 kHz and an amplitude of ±9 V. The rectified and amplified signal, after passage through the detection cell, is recorded by a 24 bit A/D converter Panther 1000, operated by the Ecomac 0.97 software (Ecom, s.r.o., Czech Republic).
The switching of the sampling valve and the starting of the piezoelectric pump during sample introduction and starting of the data registration are operated, through simple interface circuits, by a computer equipped with a PCI-6034E universal measuring card and a program created in the LabView (National Instruments, USA).
Chemicals and samples
The stock solutions of the saccharides tested, sucrose, d-lactose monohydrate, d-fructose, d-ribose (Sigma) and d-glucose anhydrous (Fluka), with a concentration of 1,000 mg L−1, have been prepared in deionized water (Milli-Q Plus, Millipore, USA) and stored in a refrigerator. To prepare the separation electrolyte, hydroxides have been used, NaOH, KOH (Sigma), and LiOH monohydrate (Merck). The samples of high-energy drinks are commonly commercially available (for the manufacturer or supplier, see the parentheses) and have their saccharides contents specified on their labels:
-
Red Bull (Red Bull GmbH, Austria)—total sugar (sucrose+glucose), 11 g/100 mL;
-
KX Energy Stimulation Drink (Cott Beverages Ltd., UK)—total sugar (sucrose+glucose–fructose syrup), 11.1 g/100 mL;
-
Kamikaze (Tecfood, Czech Republic)—total sugar (sucrose+glucose), 11.3 g/100 mL; and
-
Burn (Coca Cola)—total sugar, 13.3 g/100 mL.
Experimental procedure
The saccharide solutions in model samples have been prepared in water. The solutions of the separation electrolyte and the samples have been filtered prior to their introduction into the sampling loop, using the Rotilabo-syringe filter, pore size, 0.45 μm (Carl Roth, Germany). The samples have been introduced without interrupting the high voltage at the electrophoretic electrodes. The electrophoretic current at 5 kV has equaled 12 μA in the NaOH and LiOH electrolytes and 13 μA in KOH. After the end of a separation, the capillary has been rinsed with the pure separation electrolyte for 30 s. The capillary has been stored overnight filled with deionized water.
Samples of energy drinks have been freed from gaseous componens by 30-min sonication and diluted with deionized water at a 1:50 ratio prior to their analysis. The determination has been based on the standard addition method, to avoid the sample matrix effect on the detector response. The standard addition has amounted to 1 g L−1 in all the analyses.
Results and discussion
The effect of the kind of the hydroxide
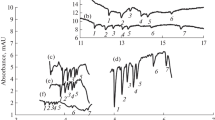
Electrophoretic separations of saccharide mixtures are performed in hydroxide solutions of pH >12, in which the saccharide anions migrate against the direction of rapid electro-osmotic flow (EOF) [12,13,20]. The saccharide zones are visualized by the C4D as negative peaks, because their mobility is lower than that of the separation electrolyte co-ion. The principal component of the separation electrolyte in the papers published so far has been sodium hydroxide. Therefore, the first step of the present study is checking, whether the hydroxide kind affects the separation. The test sample, an aqueous solution of a mixture of most common saccharides, sucrose, glucose and fructose, has been separated in KOH, NaOH, and LiOH. The appropriate electropherograms are depicted in Fig. 2; it can be seen that the hydroxide kind primarily affects the EOF velocity and thus the separation time. The EOF measured is fastest in the hydroxide whose cation migrates most rapidly, u eof (KOH) = 5.2 × 10−4 cm2 s−1 V−1; consequently, the saccharide migration times are shortest. The situation is opposite in the case of most slowly migrating cation, u eof (LiOH) = 3.7 × 10−4 cm2 s−1 V−1; however, the slower migration time permits a better analyte resolution. For example, the parameter of resolution (R) in the separation of glucose and fructose equals 1.17 ± 0.04, 1.42 ± 0.05, and 1.52 ± 0.03 for KOH, NaOH, and LiOH, respectively. It has been found that the separation efficiency, expressed in terms of the height of the theoretical plate, is affected little by the hydroxide kind. For example, the determined plate height for sucrose equals 7.0 ± 0.4, 6.3 ± 0.1, and 6.2 ± 0.3 in KOH, NaOH, and LiOH, respectively.

Electrophoretic separations of a model mixture of three saccharides in 75 mM KOH (A), NaOH (B), and LiOH (C). 1, Sucrose; 2, glucose; 3, fructose. Sample: an aqueous solution with the same saccharide concentrations of 500 mg L−1; a quartz capillary, 10 μm (i.d.), 10 cm long; distance to the detector, 8 cm; and separation voltage, 5 kV
The hydroxide cation, acting as the counter-ion to the test saccharide anions, also influences the C4D sensitivity—the detector response is highest in KOH and lowest in LiOH; for example, the ratio of the surface areas of the sucrose peaks in KOH and LiOH equals ca. 1.4. Therefore, if saccharides migrating at sufficiently different rates are to be separated, e.g., a mixture of sucrose with glucose or fructose, then KOH is the suitable electrolyte (higher detection sensitivity and a shorter separation time). If higher demands are placed on the separation, then LiOH is to be preferred (a better degree of resolution). All this holds for a capillary with an effective length of 8 cm. However, it has been found that the differences in the analyte migration times observed in NaOH and LiOH disappear in shorter capillaries. As a result and in agreement with the literature [12,13], NaOH can be considered to be a universal electrolyte, and all the following measurement of the present study have been performed in it.
Separations of saccharides in model samples
An electropherogram for the separation of five common saccharides in a capillary with an overall length of 10 cm and effective lengths of 4 cm, using the NaOH electrolyte, can be seen in Fig.3A, and the principal separation parameters found are given in Table 1. It has been found, in agreement with ref. [12], that the optimum hydroxide concentration equals 75 mM. In this medium, the test saccharides are separated within 55 s. Figure 3, curve B, demonstrates the separation in 75 mM KOH; it can be seen that the separation is very rapid, but the saccharide separation is incomplete.

An electrophoretic separation of a model saccharide mixture in 75 mM NaOH, using a quartz capillary, 10 μm (i.d.), 10 cm long, and with effective lengths of 4 cm (A). An electropherogram of this mixture in 75 mM KOH is depicted under (B). Aqueous saccharide solution: 1, sucrose; 2, lactose; 3, glucose; 4, fructose; 5, ribose, at the same concentrations of 500 mg L−1; separation voltage, 5 kV
Based on the previous results, all the following measurements have been carried out under these experimental conditions: quartz capillary, 10 μm (i.d.); total length, 10 cm; effective length, 4 cm; separation electrolyte, 75 mM NaOH; and separation voltage, 5 kV.
The concentration dependences of the saccharide peak areas have been linear within a concentration range studied; the corresponding regression parameters of the calibration straight lines are given in Table 2.
Determination of saccharides in high-energy drinks
Illustrative electrophoreograms of the saccharides contained in the test high-energy drinks with the lowest and highest contents of fructose can be seen in Fig. 4. It has been found that the calibration dependences obtained with the model solutions of the saccharides studied cannot sometimes be used in analyses of samples of energy drinks. For example, the overall saccharide contents found in the Red Bull and KX drinks were by 27 and 15 %, respectively, lower than the values specified on the drink labels. The results for the remaining drinks differed from the specified values by mere tenths or units of percent, but it is evident that the drink matrices affect the determinations. Therefore, the determinations have employed the standard addition technique. The saccharide contents determined in this way are listed in Table 3. It can be seen that the saccharide contents determined in this way are in a very good agreement with the declared values; in most cases, they are identical, within the reliability interval.

Electrophoreograms of the saccharide separations in high-energy drinks Red Bull (A) and Burn (B), diluted with deionized water at a ratio of 1:50. 1, Sucrose; 2, glucose; 3, fructose
Sucrose, glucose, and fructose have been found in all the drinks tested, even if some of them are not specified on some drink labels.
Summary of analytical parameters
The concentration dependences of the saccharide peak areas in model solutions have been linear within a concentration range tested from 50 to 1,000 mg L−1. Limit of detection (LOD) and limit of quantification (LOQ) have been computed from the relationship LOD = 3 × standard error/slope and LOQ = 10 × standard error/slope using the parameters of the calibration regression straight lines given in Table 2. The LODs and LOQs are in the range from 15 to 35 and 52 to 117 mg L−1, respectively; the LOD and LOQ values for the individual saccharides are listed in Table 2. These values are acceptable for analysis of saccharides in energy drinks.
The repeatability of the developed procedure was tested on analyses of Burn drink. For intra-day repeatability test, six samples were analyzed obtaining migration time RSD values of 1.8 % for sucrose and 2.0 % for glucose and fructose. The peak area RSD values were 2.8, 5.7, and 7.4 % for sucrose, glucose, and fructose, respectively. Analogous RSD values were obtained when the measurements were repeated in various days. Higher RSD values for the migration times and the peak areas can be expected for the laboratory prototype of the analytical apparatus used for these measurements as compared with the data obtained using a commercial equipment [12,13].
Conclusions
Electrophoretic determinations, which are commonly carried out in long capillaries of commercial instruments, can readily be performed along a separation pathway as short as 4 cm. Separations employing these short capillaries are characterized by high analysis rates, e.g., a mixture of six saccharides has been separated within a time shorter than 60 s. This is of a great importance for analyses of extensive sets of samples, required, e.g., in the control of the quality of foodstuffs. Under favorable conditions, e.g., with simple samples containing saccharides migrating with sufficiently different rates, the analysis can further be accelerated when the NaOH separation electrolyte is replaced by KOH; the time of analysis is decreased to ca. one half.
The determination is also sufficiently sensitive. The fact that the LOD values found in this work are higher than those published in some works, e.g., in ref. [13], need not adversely affect many practical applications. The contents of common saccharides are often so high, e.g., in foodstuff samples, that the samples must be diluted prior to their analysis.
The advantages of electrophoresis in short capillaries over chip electrophoresis are obvious. A commonly available separation capillary is employed, not a special separation system—a chip. The properties of standard quartz capillaries in electrophoretic separations are well known and can be suitably modified if necessary. The length and diameter of the separation space can readily be varied by altering the capillary dimensions and a damaged capillary can readily be replaced. Capillaries made of different materials, e.g., PEEK, can also be used.
References
Montero CM, Dodero MCR, Sanchez DAG, Barroso CG (2004) Chromatographia 59:15–30
Liu H, Zhang S, Yu A, Qu L, Zhao Y, Huang H, Li J (2004) Bioorg Med Chem Lett 14:2339–2344
Sesta G (2006) Apidologie 37:84–90
Heleno SA, Barros L, Sousa MJ, Martins A, Ferreira ICFR (2009) Microchem J 93:195–199
Wolfender JL (2009) Planta Med 75:719–734
El Rassi Z (1994) Carbohydrate analysis: high performance liquid chromatography and capillary electrophoresis. Elsevier, Amsterdam
Cataldi TRI, Margiotta G, Zambonin GC (1998) Food Chem 62:109–115
DIONEX (2007) http://www.dionex.com/en-us/webdocs/61831-Bro_Carbohydrates_Food_Beverage_29Aug2007_LPN1971.pdf. Accessed 8 April 2012
Soga T, Serwe M (2000) Food Chem 69:339–344
Honda S (1996) J Chromatogr A 720:337–351
Žídková J, Chmelík J (2000) Chem List 94:1093–1103
Tůma P, Málková K, Samcová E, Štulík K (2011) Anal Chim Acta 698:1–5
Carvalho AZ, da Silva JAF, do Lago CL (2003) Electrophoresis 24:2138–2143
Opekar F, Coufal P, Štulík K (2009) Chem Rev 109:4487–4499
Lauer HH, Rozing GP (2010) High performance capillary electrophoresis. Agilent Technologies, Germany
Tůma P, Opekar F, Jelínek I (2000) J Chromatogr A 883:223–230
Tůma P, Opekar F, Štulík K (2002) Electrophoresis 23:3718–3724
da Silva JAF, do Lago CL (1998) Anal Chem 70:4339–4343
Novotný M, Opekar F, Jelínek I (2005) Chem List 99:132–136
Rainelli A, Hauser PC (2005) Anal Bioanal Chem 382:789–794
Acknowledgments
Financial support from the Ministry of Education, Youth and Sports, Czech Republic, Research Project No. MSM0021620857, and the Grant Agency of the Czech Republic, grant No. P206/10/1231, is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vochyánová, B., Opekar, F., Tůma, P. et al. Rapid determinations of saccharides in high-energy drinks by short-capillary electrophoresis with contactless conductivity detection. Anal Bioanal Chem 404, 1549–1554 (2012). https://doi.org/10.1007/s00216-012-6242-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6242-x