
Abstract
Systems biology requires knowledge of the absolute amounts of proteins in order to model biological processes and simulate the effects of changes in specific model parameters. Quantification concatamers (QconCATs) are established as a method to provide multiplexed absolute peptide standards for a set of target proteins in isotope dilution standard experiments. Two or more quantotypic peptides representing each of the target proteins are concatenated into a designer gene that is metabolically labelled with stable isotopes in Escherichia coli or other cellular or cell-free systems. Co-digestion of a known amount of QconCAT with the target proteins generates a set of labelled reference peptide standards for the unlabelled analyte counterparts, and by using an appropriate mass spectrometry platform, comparison of the intensities of the peptide ratios delivers absolute quantification of the encoded peptides and in turn the target proteins for which they are surrogates. In this review, we discuss the criteria and difficulties associated with surrogate peptide selection and provide examples in the design of QconCATs for quantification of the proteins of the nuclear factor κB pathway.

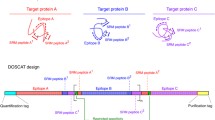
Workflow for QconCAT mediated protein quatification
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proteomics has become a quantitative science, and the need to derive accurate quantification of protein levels, whether in relative or in absolute terms, is a common goal [1]. In the now well-established discipline of systems biology, one goal is to delineate protein interaction networks and to measure protein flux within such networks. Modelling of pathways within protein networks enables predictive biology, whereby the predicted outcomes of system perturbation can be tested experimentally. Data are thus needed to parameterise models, and determination of the concentration of proteins in absolute terms, for example, as copy number per cell, is a necessity in many quantitative proteomics experiments. Many proteomics identification experiments using mass spectrometry (MS) are based on the principle of surrogacy, whereby analyte proteins are digested with a protease (often trypsin) and the MS characterisation is of the resultant peptides. Commonly, quantification is also based on the behaviour of the proteolytic fragments and can be based on label-free methods that use the inherent detectability and intensity of the peptide ion as detected in the mass spectrometer. Alternatively, the well-established approach of stable-isotope-mediated quantification, in which a stable-isotope-labelled peptide is added to a protein digest in a known amount, enables quantification of the corresponding analyte peptide and, by inference, the target protein [2–4]. Recent articles have discussed the various isotope dilution approaches for absolute quantitative proteomics and the range of methods that are available for standard generation [5, 6]; these are summarised in Table 1. In this review, we specifically discuss quantification concatamer (QconCAT)-based quantification, a molecular-biology-driven approach to the production of large numbers of highly multiplexed standards that has now been used in a range of quantification applications (Table 2).
In stable-isotope-mediated quantification of proteins, the ideal standard would be an identical protein, isotopically labelled, accurately quantified, occupying exactly the same tertiary (folded) and quaternary (complex formation) structural space and displaying an identical range of post-translational decorations. After complete proteolysis, the analyte peptides would be referenced to an identical series of standard peptides, reflecting biological or chemical modifications and partial or incomplete proteolytic release of some peptides. However, the standard proteins are usually expressed heterologously in a different organism or in a translationally active heterologous cell extract and thus may not fold to the same structure or carry the same post-translational modifications—such variances can alter the kinetics of proteolysis and could compromise the anticipation of strict stoichiometry between standard and analyte peptides. Also, the standard proteins would not be in the same quaternary structural environment as the analyte, and could lack the appropriate binding partners. The closest approximation to such protein standards are protein standards for absolute quantification (PSAQ), FlexiQuant [7–9] and variants thereof (Table 1), such as absolute stable isotope labelling by amino acids in cell culture (SILAC) [10] and full-length expressed stable-isotope-labelled proteins for quantification [11]. In an approach that does not attempt to match the standard to the analyte, protein epitope signature tags (PrESTs) produced by the Human Protein Atlas Project have been successfully deployed as a source of both non-labelled and stable-isotope-labelled peptide quantification standards in a human cancer cell line (HeLa cells) [12]. PrESTs were designed for use as antigens for antibody production and incorporate a sequence of about 100 amino acids derived from the target protein chosen for minimal homology to other proteins. PrESTs restrict the choice of quantification peptides to the epitope region of the target protein (not selected for MS performance) and could include peptides that are suboptimal for MS-based quantification, but for the human proteome, this is a very interesting approach that also allows some convergence of MS and antibody-based quantification. Even PSAQs and PrESTs are used at the peptide level, and quantification is based on standard-to-analyte ratios of peptide intensities in the mass spectrometer. For these standards, the sequence context of such peptides is identical to that of the endogenous protein, which helps equalise digestion efficiency [12] although that is only true if digestion is allowed to go to completion.
As with peptide standards for absolute quantification (AQUA peptides [13, 14]) and PSAQ or PrEST, quantification of more than one protein in a complex sample requires the expression and individual quantification of multiple standards or the chemically synthesised, stable-isotope-labelled AQUA peptides, which can be costly as the scale of quantification is increased from single or few proteins to large-scale absolute quantification studies. Moreover, for multiplexed quantification of many analytes, each standard needs to be rigorously and independently quantified. To address such issues, we developed the QconCAT approach. QconCATs are proteins encoded by synthetic genes that are concatamers of peptide internal standards (quantotypic peptides) usually with more than one peptide mapping to one target protein [15, 16]. Typically, between ten and 30 target analyte proteins are encoded in each QconCAT at a level of two quantotypic peptides per protein, leading to QconCAT proteins that are typically of mass 50–70 kDa. Because each peptide has to be in a 1:1 stoichiometry, QconCATs are ideally suited to a high level of multiplexed absolute quantification. Peptide-concatenated standards are a variant of QconCATs [17] in which short intervening sequences are used to reconstruct primary sequence context in the standard protein to match the analyte (Table 1). This review provides an overview of the construction and deployment of QconCATs—a ‘designer gene’ approach to the generation of highly multiplexed quantification standards (Table 2).
Principles of QconCAT design
Selection of quantotypic peptides
Each QconCAT is a protein, the product of a synthetic gene, that is a linear concatenation of peptides (usually tryptic but, of course, other endopeptidases of different specificities can be used). The order of the peptides is of no significance in terms of the protein primary sequence, is under the direction of the QconCAT design process and can be used to optimise digestibility or to eliminate structural features in the cognate messenger RNA that might impede translation. After expression of the labelled QconCAT, complete digestion will generate a stoichiometric set of stable-isotope-labelled standards that are formally representative of the quantity of the analyte proteins—we refer to these as quantotypic peptides. Quantotypic peptides are distinct from proteotypic peptides—peptides that are always observed for a specific protein, irrespective of whether they are suitable for quantification. Not all proteotypic peptides are quantotypic, but all quantotypic peptides are proteotypic.
The key step in QconCAT design is the selection of quantotypic peptides. There is, in principle, a great deal of freedom in QconCAT design, but as discussed below, there are multiple constraints that have to be considered. A minimum of two peptides should be selected for each protein but, of course, three peptides would provide more robust quantification, particularly in the event of disparity between individual peptide quantification data. In our experience, disparity between two peptides is attributable to incomplete digestion of the standard or analyte, or to a failure to detect the analyte, possibly because of an unanticipated post-translational modification. We classify all peptide-level quantifications as type A (good standard and analyte signals) or type B (good standard, missing analyte). Type A quantifications yield actual values; type B quantifications are taken to define the upper limit of analyte abundance and are usually set as no more than 10 % of the lowest detectable level of the standard signal. At a ratio of two peptides per analyte protein, ‘AA’ values are considered to be more reliable than ‘AB’ values. Adding a third peptide would allow an element of ‘consensus quantification’ and ‘AAB‘ values would be of higher quality than ‘AB’ values.
Presently, we lack sufficient information to simply construct a QconCAT from a definitive list of quantotypic peptides that are, a priori, known to be formally quantitatively representative of the parent protein. Until this information is in place, strategies for nomination of quantotypic peptides are either based on prior observation in MS or MS/MS experiments or based on prediction of suitable candidates in silico. Proteotypic peptide databases such as the Global Proteome Machine Database (GPMDB) [18], PeptideAtlas [19, 20] and PRIDE (proteomics identifications database) [21–23] aid peptide selection. However, these peptides are included in such databases on the basis of the frequency of observation in MS/MS studies and they are not assessed as quantitatively representative of the parent protein. There is as yet no established resource to demonstrate completeness of proteolysis, the lack of post-translational modification or, indeed, the uniqueness of the peptide and freedom from isobaric and isomeric peptides derived from other proteins. For example, many such proteotypic peptides derive from dibasic or interspersed dibasic cleavage sites, which are known to cause variable digestion and have the potential to compromise selected reaction monitoring (SRM) assays [24].
By contrast, prediction tools can ‘learn’ from global observed peptide behaviours and use this knowledge to predict candidate quantotypic peptides, albeit without taking into account post-translational modifications, for example. In addition to the known roles of peptide size, charge and hydrophobicity, peptide secondary structure is also an important factor in determining ‘detectability’ by electrospray ionisation MS [25]. A recent development in this area is the release of CONSeQuence (consensus predictor for quantotypic peptide sequence), a prediction tool for reference peptide selection for absolute quantitative proteomics based on four different machine learning approaches. CONSeQuence can be applied to lists of peptides or FASTA files containing up to 1,000 proteins via a Web interface at http://king.smith.man.ac.uk/CONSeQuence. A missed cleavage predictor (http://king.smith.man.ac.uk/mcpred/) is independently applied to a set of predicted quantotypic peptides generated by CONSeQuence; this downgrades putative peptides flanked by amino acids at their N- and C-termini that have a high probability of eliciting missed cleavages [25].
Assembly of quantotypic peptides into a QconCAT
The order of quantotypic peptides in a QconCAT is under the control of the user at the gene design phase. Because peptides in the QconCAT acquire new ‘neighbours’ in direct primary sequence juxtaposition with other standard peptides, the sequence context, and thus the digestion context, of the standard and analyte can differ, and proteolytic excision of the peptide may occur at a different rate in the QconCAT than in the analyte. It is therefore prudent to assemble the peptides in a pattern that preserves, as much as possible, local primary sequence context. For example, if an alanine residue follows the tryptic cleavage site, the corresponding quantotypic peptide could be placed adjacent to a second quantotypic peptide with an N-terminal alanine. However, consideration of the primary sequence context of peptides nominated as standard peptides has to extend beyond the immediate neighbouring residue. In fact, the efficiency of proteolytic cleavage is influenced by residues at least three positions distal to the cleavage site [26]. A preponderance of acidic amino acids can prevent complete digestion, and dibasic sites, or interspersed dibasic sites [24], can lead to miscleavages that cannot be resolved by extended incubation with trypsin. One solution is the insertion of short peptide sequences of three to four amino acids in length, juxtaposed between each quantotypic peptide, to mimic the primary sequence context of the analyte protein [17, 27]. Although it might be argued that partial digestion is not a problem if the analyte and standard show the same extent of cleavage, in the QconCAT workflow it is essential that the digestion of both the analyte and the standard is complete. This eliminates error in the quantification method caused by differences in the proteolysis kinetics and emphasises an ‘end-point’ quantification based on the products of digestion. Judicious selection of peptides can reduce miscleavage problems, and the determination of the digestion kinetics of both QconCAT and analyte peptides monitored by MS analysis of isotope ratios should enable assessment of the efficiency of digestion and highlight any problematic peptides [24].
Because the QconCAT gene has to be synthesised de novo, it also provides the opportunity to introduce additional features. At the N-terminus, a short sacrificial peptide provides the initiator methionine and also protects the N-terminus of the first true quantotypic peptide (which is often a QconCAT quantification standard) from exoproteolytic trimming. Glufibrinopeptide B (Glufib) is routinely used in this position, enabling quantification of the stable-isotope-labelled QconCAT by reference to an accurately quantified, unlabelled Glufib peptide standard. At the C-terminus, a sequence variant of Glufib is incorporated, allowing two-point quantification and, if required, confirmation that the QconCAT is intact. The extreme C-terminus encodes a hexahistidine purification tag (His-tag) that is used for purification of QconCATs on nickel affinity columns should that be required.
Expression of labelled QconCATs
QconCATs are the products of synthetic genes optimised for heterologous expression in Escherichia coli [15, 16]. Codon optimisation and the design of RNA secondary structure to minimise the likelihood of hairpin loops that impede successful translation have to be taken into consideration as messenger RNA folding near the ribosomal binding site and the associated rates of translation initiation play a predominant role in the expression levels of individual genes. Moreover, codon bias influences global translation efficiency and cellular fitness, so QconCAT genes are encoded with high-abundance codons [28]. QconCATs are designed to generate a stoichiometric set of quantification peptides, and the selection criteria include a limited peptide mass range and avoidance of specific amino acids and sequences. It is possible, therefore, that the properties of QconCATs are sufficiently different from those of native proteins to compromise expression. To test this, we performed a principal component analysis of approximately 100 QconCATs (made to quantify yeast proteins [29]), approximately 100 yeast proteins (selected at random from the Saccharomyces cerevisiae proteome) and approximately 100 E. coli proteins. The two-dimensional projections of the three-dimensional loadings plot of the data set (Fig. 1a) indicate that the three clusters (E. coli, yeast and QconCATs) are readily separated by such an analysis. However, if the amino acid composition data are modified to eliminate lysine, arginine and methionine, reflecting important drivers in QconCAT design (Fig. 1b), the three data groups converge such that is not possible to distinguish the QconCATs from the yeast proteins, although the E. coli group remain slightly discriminable. These data should not be overinterpreted, but serve to indicate that, to a first approximation, QconCATs resemble native proteins in composition. The pronounced suppression of lysine, arginine and methionine might, however, be sufficient to enforce insolubility on most QconCATs, a factor that aids purification. The expression of heterologous proteins in E. coli is not always straightforward [30], but expression of QconCATs may be less problematic than that of natural proteins since there is no requirement for functional folding or solubility. In practice, the bacterial expression of the codon-optimised gene is sufficiently strong that the QconCAT becomes the major protein band in a broken cell extract resolved by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), and the overexpressed QconCATs usually aggregate as inclusion bodies. The abundance of the QconCAT in the inclusion bodies is often sufficiently high so as not to compromise analytical complexity, rendering His-tag-mediated purification unnecessary.

Principal component analysis of quantification concatamers (QconCATs) and natural proteins. One hundred and eight QconCATs (Q) designed to quantify proteins from Saccharomyces cerevisiae and an equivalent number of native protein sequences from S. cerevisiae (Y) and Escherichia coli (E) were analysed for fractional amino acid composition. The composition data were then used as input to a principal component analysis, and the projection planes of the three component model scores, together with the error analysis of the score values are displayed. a Analysis using all amino acids. b The same analysis but with the fractional abundances of the amino acids lysine, arginine and methionine removed from the input data. Each group of input proteins are represented by individual values connected to the group centroid (mean value). PC principal component

Selection of quantotypic peptides. For each member of the nuclear factor κB (NF-κB) protein set, the number of possible quantotypic candidate peptides available for each target protein for possible inclusion in a QconCAT is highlighted at the left of each cluster. The first column of each cluster represents the total number of tryptic limit peptides. A size filter of 800–3,000 Da was then applied, followed by composition filters—sequence contexts NG (asparagine prone to deamidation), DP (possible non-enzymatic cleavage), N-terminal glutamine (variable pyroglutamic acid formation) and methionine (variable oxidation). The remaining peptides were screened for known sites of post-translational modification (PTM). Finally, the local peptide sequence context within the target protein was examined. Peptides within dibasic sequence contexts that could lead to alternative products and peptides known to impede the action of trypsin such as -EE- were also filtered out, leaving a reduced set of putatively quantotypic peptides at the right of each cluster

Tryptic peptide maps of a RelA and b IκBα. The peptide maps show details of tryptic peptides larger than 800 Da annotated with monoisotopic [M + H]+ masses (including adjustment for cysteine carbamidomethylation modification). Peptides within a size range suitable for quantotypic peptide selection (800–3,000 Da) are represented by boxes. KAc lysine acetylation, pT phosphothreonine, pS phosphoserine, pY phosphotyrosine

Induction and purification of NF-κB QconCAT1. a Representation of NF-κB QconCAT1 highlighting the disposition of each peptide and the protein upon which it reports. b The QconCAT gene was supplied in a pET21b plasmid and transformed into E. coli BL21DE3 cells, and expression was monitored over 4 h. c Inclusion bodies were solubilised and purified by nickel affinity chromatography. Two major truncation products were observed and mapped to fragments of the QconCAT. d Bacterial cell lysate samples from the induction time course (b) were resolved by sodium dodecyl sulphate polyacrylamide gel electrophoresis, transferred to nitrocellulose and the blot was probed with an anti-hexahistidine tag (His-tag) monoclonal antibody. e Imidazole-eluted fractions from His-tag-purified NF-κB QconCAT2 and NF-κB QconCAT3
Ideally, the QconCAT should be fully labelled with the stable isotope amino acid(s). It is worth noting that the dynamic range of the quantification experiment is largely controlled by the efficiency of labelling of the QconCAT. If a ‘heavy’ standard contains 1 % ‘light’ unlabelled standard, it is not feasible to quantify an analyte at less than about 5 % of the standard signal. If 10 % of the QconCAT were synthesised in an unlabelled form, the dynamic range would reduce to less than one order of magnitude. For this reason alone, complete labelling should be sought. In practice, this is not achievable. First, the isotopic purity of the commercially available amino acids used in the medium is usually marginally (1–2 %) less than 100 %. Second, unless strains that are auxotrophic for the labelling amino acids are used, there is the possibility that the cell will synthesise unlabelled amino acid and reduce the labelling efficiency, although in our experience this is rarely a problem provided that care is given to the choice of growth medium—for example, a QconCAT expressed in E. coli in SILAC cell culture medium (Dulbecco’s modified Eagle medium, designed for mammalian cells) resulted in virtually complete isotopic labelling [31]. Several groups have employed cell-free expression systems [7, 10, 27, 32] to reduce metabolism of stable-isotope-labelled precursors. A tightly controlled expression system such as the T7/pET system [33] directs most of the synthetic effort to the synthesis of the heterologous protein, and labelling is both efficient and as complete as can be expected. QconCATs are expressed in minimal media containing the stable-isotope-labelled amino acids of choice (usually [13C6]lysine and [13C6]arginine so that all tryptic peptides contain one instance of the label-derived mass offset) and the efficiency of labelling is greater than 98-99 % (Fig. 5). In most instances the expression is sufficiently high that the QconCAT becomes the dominant protein band on a gel of the broken cell pellet within 1 h of induction [29, 34].

Efficiency of labelling of QconCAT peptides. An in-solution tryptic digest of both unlabelled and [13C6]arginine- and [13C6]lysine-labelled NF-κB QconCAT3 was analysed by matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry. The isotopomer peak profiles of a quantotypic peptide selected for the quantification of c-Rel show the labelled peptide superimposed on the spectrum of the unlabelled peptide. The lack of unlabelled peptide in the stable-isotope-labelled QconCAT is evident
QconCATs are frequently obtained in multi-milligram quantities from 200-mL bacterial cultures [29, 34]. For a typical QconCAT (mass approximately 60 kDa), this is equivalent to about 100 nmol of standard, an amount that should be seen in the context of a typical SRM assay that requires 1 fmol (or less) of standard! Indeed, we have rarely needed to express a QconCAT more than once. From the accumulated experience of over 120 QconCATs, with a single exception, all of these artificial proteins have been expressed in inclusion bodies—a positive feature, since centrifugal recovery of inclusion bodies represents an immediate tenfold purification step. The inclusion bodies are then dissolved in chaotropes prior to affinity chromatography, a second assurance that ensures that there are no higher-order structure constraints to impede proteolysis. We have successfully used inclusion body preparations, dissolved in the digestion enhancer detergent RapiGest, without further purification. The degree of E. coli contamination in these preparations is low, and is effectively rendered ‘invisible’ by virtue of the specificity of the SRM assay. A soluble QconCAT has been obtained as a glutathione S-transferase (GST) fusion [31]. Whether GST purification tags can routinely predispose the fusion protein to be expressed in soluble form awaits further testing. Moreover, the GST tag (26 kDa) is substantially larger than a His-tag, increasing the overall size of the QconCAT. It is possible that soluble QconCATs might be more susceptible to endogenous proteolysis.
Deployment of QconCATs
Any quantitative study is critically dependent on a workflow in which there can be no differential loss of analyte and standard. The workflow can be broken down into analyte preparation, standard preparation, proteolysis and MS analysis. To illustrate the critical importance of each step, we refer to a recent quantification study of 27 proteins in the glycolytic pathway of S. cerevisiae, covering a dynamic range of 14,000 to ten million molecules per cell [35]. Two quantotypic peptides per target protein were used, resulting in a final QconCAT protein of average mass 87.8 kDa, including the sacrificial N-terminal segment and purification tag. To quantify the more abundant proteins, extracted precursor ion chromatograms were used to generate peak areas for the analyte and standard peptides in full MS/MS runs and a targeted SRM approach was employed for the lower-abundance proteins. A rigorous assessment of protein recovery and digestion was made. Yeast cells were disrupted using glass beads for five successive disruption cycles. Two rounds of protein extraction recovered only 50-68 % of protein and it was necessary to perform an additional three rounds for complete extraction of the protein pool. At each extraction stage the supernatant fraction was removed and co-digested with stable-isotope-labelled QconCAT for comparison of peptide ratios. A simple centrifugation clarification step is unlikely to create a soluble fraction that contains all soluble protein. For this reason, we now prefer to use unfractionated, uncentrifuged broken-cell biological samples as the analyte input material [29, 35].
A critical element of QconCAT deployment is the stage in the workflow at which the analyte protein mixture (inevitably a mixture, because the QconCAT is inherently multiplexed) and standard are combined. If a protein mixture is enriched or prefractionated, then QconCAT quantification can quantify proteins in the recovered fraction, but these might not reflect absolute cellular abundances. This approach can be of value, for example, in determination of protein stoichiometry in multiprotein complexes [31, 36]. Similarly, quantification of material derived from a gel slice would need to be assessed for errors derived from incomplete excision or digestion of the protein in the gel slice. By the same argument, QconCATs do not occupy the same physicochemical space as the analyte proteins, and cannot be assumed to fractionate, chromatograph or co-precipitate with the analyte. However, if the QconCAT and analyte are digested to completion, any steps that fractionate or concentrate peptides can be assumed to elicit the same behaviour in the standard and the analyte, and the true quantification is sustained through the sample workup and the transition from protein space to peptide space. QconCAT approaches are thus robust to two-dimensional chromatography or prior isoelectric fractionation of peptides, and an optimal workflow would be based on an early digestion step and subsequent fractionation and analysis of peptides.
To obtain accurate quantification data, peptides must be completely released from both the parent protein and the QconCAT. Optimisation of analyte digestion conditions is a crucial preliminary step in any proteomics experiment that has the goal of absolute quantification. A major determinant of the rate of proteolysis of native proteins is higher-order structure. Proteins that are tightly folded, and especially those with a high percentage of β-sheet, demonstrate intrinsic resistance to proteolysis [37–39]. Our studies on the rate and completeness of proteolysis in QconCAT experiments have demonstrated that QconCATs are digested rapidly and completely (within a few minutes) and unless appropriate denaturation steps are used, analyte digestion can be substantially slower. In proteomics experiments it is therefore critical to remove such barriers to digestion by reduction of disulphide bonds, by blocking disulphide re-formation with alkylating agents and by the use of chaotropes to unfold proteins (urea, guanidinium hydrochloride, acetonitrile) or heat denaturation (exceptionally, even these treatments in combination). We routinely employ a digestion enhancer detergent (RapiGest) to minimise complications from incomplete proteolysis. A customised FASTA database containing the sequence of the recombinant QconCAT protein is used to facilitate detection of quantotypic peptides and any possible miscleavages. In the glycolysis study described above, the kinetics of release of peptides from the combined extractions following tryptic digestion was monitored by comparison of the ratio of labelled to unlabelled signal over time. After 250 min of digestion, the relative proportion of analyte to standard signal reached a stable plateau, consistent with complete proteolysis [35].
Case study: design of QconCATs for absolute quantification of components of the nuclear factor κB pathway
To illustrate the challenges and complexities of QconCAT design, we describe the design of standards for the nuclear factor κB (NF-κB) pathway. NF-κB transcription factors activate more than 300 genes responsible for the cellular response to challenge mediating inflammation, immunity and cell survival. There are five mammalian reticuloendotheliosis (Rel) family /NF-κB proteins, and they belong to two groups. The first group consists of RelA (also known as p65), c-Rel and RelB, synthesised in their mature forms. The second group includes NF-κB1 (also known as p105) and NF-κB2 (also known as p100), which are proteolytically processed to produce mature p50 and p52 proteins, respectively. The activity of NF-κB is tightly regulated, the protein being kept inactive in most cells and sequestered in the cytoplasm by members of the IκB family of inhibitory proteins (IκBα, IκBβ, IκBε). In response to stimulation, IκB proteins are polyubiquitylated and rapidly degraded by the proteasome, permitting NF-κB to enter the nucleus and activate a wide spectrum of target genes. Extensive systems modelling of this model has led to a formal description of the nuclear/cytoplasmic oscillation of p65 after stimulation, and to extend the scope of the model, accurate numbers of each pathway member, in copies per cell, were required. The ten NF-κB proteins for which quantitative data were required were RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105), NF-κB2 (p52/p100), IκBα, IκBβ, IκBε. Three QconCATs have been designed and expressed for this pathway. The intention of this review is not to provide the detailed quantification results, but to illustrate the process of QconCAT design. The three QconCATs span several years of research and illustrate the development of our understanding of their design and usage. Lastly, the two NF-κB precursor proteins NF-κB1 (p105) and NF-κB2 (p100) are cleaved in vivo. NF-κB1 (p105) is processed to p50 by the action of the 20S proteasome degrading the entire C-terminal end of the molecule in a ubiquitin-independent manner [40]. However, p52 is co-translationally generated from p100 in response to non-canonical signalling events [41, 42]. Two peptides were included in the QconCATs to span the biological cleavage sites within p105 and p100 and, thus, quantify the uncleaved peptide. This would permit the extent of cleavage and cellular survival of the proteolytic products to be assessed by difference.
Design of NF-κB QconCATs
For each target protein, we performed tryptic digests in silico, and identified peptide candidates for quantification. After the application of a series of filters, the number of candidate peptides useable in principle for each protein was dramatically reduced (Fig. 2). To illustrate the difficulty of selection of appropriate quantotypic peptides, peptide maps of two proteins selected for inclusion in the QconCAT for quantification of NF-κB proteins RelA (or p65) and IκBα are shown in Fig. 3. The peptide map of RelA (60.2 kDa) illustrates that approximately one third of the protein is encompassed by T47, a 17-kDa segment devoid of tryptic cleavage sites. Nineteen tryptic peptides in the size range 800–3,000 Da were initial candidates; however, three of the peptides contained one or more known sites of post-translational modification. Twelve peptides remained before other selection criteria such as sequence context were considered. Smaller proteins such as IκBα (34.8 kDa) generated fewer tryptic peptides; from the eight peptides satisfying the above-mentioned size filter, two were subject to post-translational modification so were discounted. Six peptides remained for selection by sequence criteria, exemplifying once again the high attrition rate experienced with peptide selection.
For the ten proteins from the NF-κB pathway, two quantification peptides per protein were selected, on the basis of their observed frequency in PeptideAtlas and GPMDB, and for those proteins for which no peptides were observed, the PeptideSieve [43] score was used as a selection guide taking other criteria into account. For eight of the ten proteins encoded in the QconCAT, at least one of the two selected quantotypic peptides had been observed in either PeptideAtlas or GPMDB. The exception to this was IκBα, for which the two peptides observed were too large to be optimal for targeted multiple reaction monitoring (MRM) experiments (more than 30 amino acids; T16, Fig. 3b). A total of five peptides were selected for the quantification of p50/p105, two peptides for p50, two peptides for the IκBγ moiety of the protein and a tryptic peptide spanning the site of proteolytic processing (Table 3). In addition, four peptides were included for the quantification of p52/p100 for both the N-terminal moiety processed to p52 and the IκBδ moiety. Of the peptides present in proteotypic peptide databases, several were discounted because of the presence of methionine (the variable oxidation can ‘split’ the quantotypic signal) or because of ‘difficult’ cleavage contexts [24].
Expression and characterisation of NF-κB QconCATs
QconCATs are novel proteins, and their behaviour is unpredictable. In rare instances (two occurrences from more than 150 QconCATs) a full-length QconCAT has not been obtained after purification. This occurred with the first iteration of the first QconCAT (NF-κB QconCAT1; Fig. 4a). The NF-κB QconCAT1 was induced over 5 h—after 1 h the protein was expressed strongly (Fig. 4b). The purification protocol started with gentle lysis of the bacterial cell pellets in a proprietary detergent-based solution containing a protease inhibitor cocktail [34]. The QconCAT, present in inclusion bodies, was then solubilised in buffers containing 6 M guanidinium hydrochloride and purified by metal affinity chromatography (Fig. 4b). Although a full-length QconCAT was obtained, two truncated QconCAT fragments, both lacking N-terminal peptides, were present—verified by matrix-assisted laser desorption/ionisation time of flight (MALDI-ToF) MS. An additional band was detected in the bacterial cell lysates and the smaller of the two truncated QconCAT bands was just visible 4-5 h after induction (Fig. 4c). Since cleavage of the QconCAT was at such an early stage in the purification procedure and protease inhibitors were ineffective in preventing the adventitious proteolysis, the peptides containing the cleavage sites were identified by a combination of MALDI-ToF MS and MS/MS (RelB peptide LTDGVCSEP↓LPFTYLPR and p100 peptide TFAGNTPLH↓LAAGLGYPTLTR; arrows indicate the sites of cleavage), and these proteolytically susceptible peptides were replaced by new quantotypic peptides in a second iteration of the QconCAT (NF-κB QconCAT2). This was expressed at high yield with no evidence of proteolysis (Fig. 4e). During the research programme, the development of CONSeQuence [25] gave an opportunity to compare predicted peptides with those used in the QconCATs. We therefore designed a third QconCAT on the basis of CONSeQuence prediction. This QconCAT was also expressed at high levels (Fig. 4e).
High-throughput methods for the empirical discovery of optimal proteotypic peptides using in vitro synthesised proteins can also be a useful screening approach. Such methods utilising tagged complementary DNA clones have been employed with human transcription factors for the discovery of fragment ions for targeted MRM experiments [44]. As part of this study, the relative intensities of all tryptic peptides generated from in vitro synthesised p50/p105 were compared to identify the most sensitive proxy to act as a surrogate for the target protein. Twenty-three peptides were rated with a high peptide score and these were split almost equally between the p50 and IκBγ sections of the protein. However, from the 23 peptides, four contained residues that were sites of post-translational modification. An additional seven peptides contained either methionine or N-terminal glutamine residues that under variable oxidation or pyroglutamic acid modification have the potential to diminish signal strength in SRM assays. A further three peptides were derived from dibasic cleavage sites that increase the likelihood of miscleavage upon tryptic digestion. Other selection criteria flagged two other peptides with unfavourable sequences (‘NG’ conducive to deamidation; EE impeding trypsin digestion), reducing the original list further. Of the remaining seven peptides, one peptide (LGLGILNNAFR) was a standard peptide in NF-κB QconCAT1 and was a proteotypic peptide listed in GPMDB; three other peptides were encoded in NF-κB QconCAT3.
Stable-isotope-labelled standard peptides were synthesised to quantify proteins in the NF-κB pathway for the quantification of proteins in human myeloma cell lines and their drug-resistant counterparts [45]. In some instances one peptide was developed for quantification, although additional peptides were monitored in subsequent biological experiments in order to provide additional confirmation of changes in expression. Protein extracted from approximately 40,000 cells (8 μg) was separated by SDS-PAGE, regions containing the proteins of interest were digested in gel and the stable-isotope-labelled standard peptides were added to the digest prior to MRM analysis. Because of the possibility of incomplete digestion and peptide recovery from the gel, absolute quantification by liquid chromatography–MRM would establish a minimum level of expression. The limit of quantification for this experiment ranged from 100 to 5,000 amol, based on the linear range of the calibration curve for each peptide standard. This highlights the analytical constraints of direct liquid chromatography–SRM analysis of trypsin-digested cell lysates, where the analytical on-column cell load is typically in the region of 2,000-4,000 human cells (500 ng protein equivalent analytical column load), compared with the 40,000-cell protein load fractionated by SDS-PAGE—a tenfold higher protein load that lowers the achievable limit of quantification.
Conclusions
The need for protein quantification, of deeper and deeper sensitivity, has invoked a need for more and more sophisticated quantification approaches, and new strategies in standard generation. A major step forward has been the development of methods for de novo gene synthesis—the ability to create designer genes, optimally tuned to the anticipated output, makes the subsequent steps rather straightforward. Expression of the products of such designer genes is increasingly facile by highly efficient in vivo or in vitro expression systems. Moreover, quantitative proteomics requires relatively small amounts of these proteins and small-scale synthesis is acceptable. Although synthesis and expression of the genes are both straightforward, the same cannot be said about the initial design phase. Optimal selection of truly quantotypic peptides can still be variably successful, and the combination of better prediction tools and increasing bodies of observational data can only assist in this regard. For a limited number of proteins it might be advisable to express each protein in vitro and assess which peptides are most consistent in the quantification data they generate—these are likely to be the optimal candidates for quantotypic peptides. Also, as we gain increasing information on sites of post-translational modifications, we will learn how to factor these into or out of peptide selection. We maintain that partial digestion is unacceptable for a rigorous quantitative study, since this is potentially variable. Thus, we must select peptides and assemble them judiciously to meet the needs of the proteolytic step. Finally, ordering of peptides in a QconCAT can ensure complete cleavage, potentially by the inclusion of naïve sequence-intervening short fragments [17].
Finally, the ease of in vitro gene synthesis means that the generation of artificial proteins and concatamers can extend beyond QconCATs. To be able to correlate proteome data sets from diverse sources there is a growing requirement for optimisation and standardisation of all commonly used instrumentation platforms for proteomics. The QCAL QconCAT (52.2 kDa), comprising 22 peptides, ranging from approximately 410 to 3,100 Da, was designed to evaluate peptide separation by reversed-phase chromatography, to facilitate the assessment and optimisation of instrument resolution and to evaluate the linearity of signal detection in different MS instruments, including MALDI-ToF, electrospray ionisation and Fourier transform ion cyclotron resonance instruments [46]. We can anticipate the emergence of other classes of ‘designer proteins’ in proteomics.
Abbreviations
- Glufib:
-
Glufibrinopeptide B
- GPMDB:
-
Global Proteome Machine Database
- GST:
-
Glutathione S-transferase
- His-tag:
-
Hexahistidine purification tag
- MALDI-ToF:
-
Matrix-assisted laser desorption/ionisation time of flight
- MRM:
-
Multiple reaction monitoring
- MS:
-
Mass spectrometry
- NF-κB:
-
Nuclear factor κB
- PrEST:
-
Protein epitope signature tag
- PSAQ:
-
Protein standards for absolute quantification
- QconCAT:
-
Quantification concatamer
- Rel:
-
Reticuloendotheliosis
- SDS-PAGE:
-
Sodium dodecyl sulphate polyacrylamide gel electrophoresis
- SILAC:
-
Stable isotope labelling by amino acids in cell culture
- SRM:
-
Selected reaction monitoring
References
Elliott MH, Smith DS, Parker CE, Borchers C (2009) J Mass Spectrom 44:1637–1660
Barnidge DR, Dratz EA, Martin T, Bonilla LE, Moran LB, Lindall A (2003) Anal Chem 75:445–451
Barr JR, Maggio VL, Patterson DGJ et al (1996) Clin Chem 42:1676–1682
Bettmer J (2010) Anal Bioanal Chem 397:3495–3502
Brun V, Masselon C, Garin J, Dupuis A (2009) J Proteomics 72:740–749
Kito K, Ito T (2008) Curr Genomics 9:263–274
Brun V, Dupuis A, Adrait A et al (2007) Mol Cell Proteomics 6:2139–2149
Dupuis A, Hennekinne JA, Garin J, Brun V (2008) Proteomics 8:4633–4636
Hennekinne JA, Brun V, De Buyser ML, Dupuis A, Ostyn A, Dragacci S (2009) Appl Environ Microbiol 75:882–884
Hanke S, Besir H, Oesterhelt D, Mann M (2008) J Proteome Res 7:1118–1130
Singh S, Springer M, Steen J, Kirschner MW, Steen H (2009) J Proteome Res 8:2201–2210
Zeiler M, Straube WL, Lundberg E, Uhlen M, Mann M (2012) Mol Cell Proteomics 11:O111.009613
Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP (2003) Proc Natl Acad Sci USA 100:6940–6945
Kirkpatrick DS, Gerber SA, Gygi SP (2005) Methods 35:265–273
Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ (2005) Nat Methods 2:587–589
Pratt JM, Simpson DM, Doherty MK, Rivers J, Gaskell SJ, Beynon RJ (2006) Nat Protoc 1:1029–1043
Kito K, Ota K, Fujita T, Ito T (2007) J Proteome Res 6:792–800
Craig R, Cortens JP, Beavis RC (2004) J Proteome Res 3:1234–1242
Desiere F, Deutsch EW, Nesvizhskii AI et al (2005) Genome Biol 6:R9
Desiere F, Deutsch EW, King NL et al (2006) Nucleic Acids Res 34:D655–D658
Vizcaino JA, Cote R, Reisinger F et al (2009) Proteomics 9:4276–4283
Vizcaino JA, Reisinger F, Cote R, Martens L (2010) Curr Protoc Protein Sci Unit 25.4
Vizcaino JA, Reisinger F, Cote R, Martens L (2011) Methods Mol Biol 696:93–105
Brownridge P, Beynon RJ (2011) Methods 54:351–360
Eyers CE, Lawless C, Wedge DC, Lau KW, Gaskell SJ, Hubbard SJ (2011) Mol Cell Proteomics 10:M110.003384
Siepen JA, Keevil EJ, Knight D, Hubbard SJ (2007) J Proteome Res 6:399–408
Nanavati D, Gucek M, Milne JL, Subramaniam S, Markey SP (2008) Mol Cell Proteomics 7:442–447
Kudla G, Murray AW, Tollervey D, Plotkin JB (2009) Science 324:255–258
Brownridge P, Holman SW, Gaskell SJ et al (2011) Proteomics 11:2957–2970
Benita Y, Wise MJ, Lok MC, Humphery-Smith I, Oosting RS (2006) Mol Cell Proteomics 5:1567–1580
Ding C, Li Y, Kim BJ et al (2011) J Proteome Res 10:3652–3659
Mirzaei H, McBee JK, Watts J, Aebersold R (2008) Mol Cell Proteomics 7:813–823
Studier FW (1991) J Mol Biol 219:37–44
Brownridge P, Harman VM, Simpson DM, Beynon RJ (2012) Methods Mol Biol (in press)
Carroll KM, Simpson DM, Eyers CE et al (2011) Mol Cell Proteomics 10:M111.007633
Olinares PD, Kim J, Davis JI, van Wijk KJ (2011) Plant Cell 23:2348–2361
Hubbard SJ (1998) Biochim Biophys Acta 1382:191–206
Hubbard SJ, Beynon RJ, Thornton JM (1998) Protein Eng 11:349–359
Wu C, Robertson DH, Hubbard SJ, Gaskell SJ, Beynon RJ (1999) J Biol Chem 274:1108–1115
Moorthy AK, Savinova OV, Ho JQ, Wang VY, Vu D, Ghosh G (2006) EMBO J 25:1945–1956
Shih VF, Tsui R, Caldwell A, Hoffmann A (2011) Cell Res 21:86–102
Sun SC (2011) Cell Res 21:71–85
Mallick P, Schirle M, Chen SS et al (2007) Nat Biotechnol 25:125–131
Stergachis AB, MacLean B, Lee K, Stamatoyannopoulos JA, MacCoss MJ (2011) Nat Methods 8:1041–1043
Xiang Y, Remily-Wood ER, Oliveira V et al (2011) Mol Cell Proteomics 10:M110.005520
Eyers CE, Simpson DM, Wong SC, Beynon RJ, Gaskell SJ (2008) J Am Soc Mass Spectrom 19:1275–1280
Anderson L, Hunter CL (2006) Mol Cell Proteomics 5:573–588
Lebert D, Dupuis A, Garin J, Bruley C, Brun V (2011) Methods Mol Biol 753:93–115
Winter D, Seidler J, Kugelstadt D, Derrer B, Kappes B, Lehmann WD (2010) Proteomics 10:1510–1514
Zinn N, Winter D, Lehmann WD (2010) Anal Chem 82:2334–2340
Remily-Wood ER, Koomen JM (2012) J Mass Spectrom 47:188–194
Austin RJ, Chang DK, Holstein CA et al (2012) Proteomics. doi:10.1002/pmic.201100626
Bislev SL, Kusebauch U, Codrea MC et al (2012) J Proteome Res 11:1832–1843
Southworth PM, Hyde JE, Sims PF (2011) Malar J 10:315
Castro-Borges W, Simpson DM, Dowle A et al (2011) J Proteomics 74:1519–1533
Johnson H, Eyers CE, Eyers PA, Beynon RJ, Gaskell SJ (2009) J Am Soc Mass Spectrom 20:2211–2220
Rivers J, Hughes C, McKenna T et al (2011) PLoS One 6:e28902
Rivers J, Simpson DM, Robertson DH, Gaskell SJ, Beynon RJ (2007) Mol Cell Proteomics 6:1416–1427
Acknowledgments
D.M.S. and R.J.B. gratefully acknowledge funding from the Biotechnology and Biological Sciences Research Council (BBSRC) Systems Approach to Biological Research (SABR) grant BB/F005938/1 and BBSRC LOLA grant BB/G009112/1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical issue Quantitative Mass Spectrometry in Proteomics with guest editors Bernhard Küster and Marcus Bantscheff.
Rights and permissions
About this article
Cite this article
Simpson, D.M., Beynon, R.J. QconCATs: design and expression of concatenated protein standards for multiplexed protein quantification. Anal Bioanal Chem 404, 977–989 (2012). https://doi.org/10.1007/s00216-012-6230-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6230-1