
Abstract
Algal blooms are well-known sources of acute toxic agents that can be lethal to aquatic organisms. However, one such toxin, β-N-methylamino-l-alanine (BMAA) is also believed to cause amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease. The detection and identification of BMAA in natural samples were challenging until the recent introduction of reliable methods. However, the issue of potential interference from unknown isomers of BMAA present in samples has not yet been thoroughly investigated. Based on a systematic database search, we generated a list of all theoretical BMAA structural isomers, which was subsequently narrowed down to seven possible interfering compounds for further consideration. The seven possible candidates satisfied the requirements of chemical stability and also shared important structural domains with BMAA. Two of the candidates, 2,4-diaminobutyric acid (DAB) and N-(2-aminoethyl) glycine (AEG) have recently been studied in the context of BMAA. A further isomer, β-amino-N-methyl-alanine (BAMA), has to be considered because it can potentially yield the fragment ion, which is diagnostic for BMAA. Here, we report the synthesis and analysis of BAMA, together with AEG, DAB, and other isomers that are of interest in the separation and detection of BMAA in biological samples by using either high-performance liquid chromatography or ultra-high-performance liquid chromatography coupled with tandem mass spectrometry. We detected for the first time BAMA in blue mussel and oyster samples. This work extends the previously developed liquid chromatography–tandem mass spectrometry platform Spacil et al. (Analyst 135:127, 2010) to allow BMAA isomers to be distinguished, improving the detection and identification of this important amino acid.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The neurotoxic non-proteinogenic amino acid β-N-methylamino-l-alanine (BMAA) was first reported to be produced by symbiotic cyanobacteria (Nostoc sp.) in the coralloid roots of the cycad trees (Cycas micronesica Hill) [2]. A subsequent report indicated that BMAA can also be produced by 95 % of cyanobacterial genera in freshwater, brackish water, and marine ecosystems [3]. Further reports have shown that BMAA can be detected in other cyanobacterial samples [4–8]. However, controversy remains regarding the distribution of BMAA in nature and its apparent harmful effects based on animal model studies as well as clinical results on patients. Therefore, the development of a robust and reliable analytical method is critical to enable confident identification of BMAA. A variety of methods have been developed based on liquid chromatography-fluorescence detection (LC-FLD), liquid chromatography–mass spectrometry (LC-MS), liquid chromatography–tandem mass spectrometry (LC-MS/MS) or gas chromatography–mass spectrometry (GC-MS), two-dimensional gas chromatography-time-of-flight/mass spectrometry to detect either underivatized BMAA [9–11] or BMAA derivatives using reagents such as 9-fluorenylmethyl chloroformate [12], ethyl chloroformate [13–15], or 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) [1,5–7,16–20]. However, conflicting results have been obtained using different methods. For instance, high quantities of BMAA were detected in cyanobacteria from across the world using LC-FLD and LC-MS methods based on derivatization [3], whereas other studies using an LC-MS/MS method and not employing derivatizaiton failed to detect BMAA in large sets of cyanobacteria [11,21]. These discrepancies may be due to differences in the biological materials (cultured under different laboratory conditions or collected from different ecosystems), or they may be directly related to the inherent properties of the analytical method, such as insufficient sensitivity and/or selectivity for unambiguous detection of BMAA in the complex sample matrices. Improved selectivity has been achieved using techniques based on tandem mass spectrometry (MS/MS) to detect BMAA diagnostic product ions at m/z 88 (for native BMAA) or m/z 258 (for AQC derivates of BMAA). The latter approach developed previously by our group [1], uses multiple features for confident BMAA identification: (1) reversed-phase liquid chromatographic separation, (2) MS/MS detection by selected reaction monitoring (SRM) of diagnostic product ions (459 > 258 for BMAA and 459 > 188 for 2,4-diaminobutyric acid (DAB)), and (3) the consistent ratio of peak areas for two product ions (119/258 for BMAA and 119/188 for DAB). Our recent analysis revealed that the ratio of SRM ion traces at m/z 119/258 for cyanobacterial samples does not always correspond to the ratio measured for standard solutions, which invokes the question of possible chemical interferences by BMAA isomers.
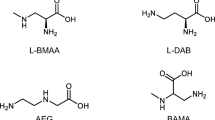
We performed a database search (Scifinder, PubMed) and identified about 260 theoretical structural isomers of BMAA. The list was narrowed down dramatically by imposing certain restrictions namely: (1) the isomers which did not contain two amino groups (either primary or secondary) were excluded as this is a required feature to generate doubly labeled AQC analogues of BMAA; (2) the isomers containing carbamic acid, carbamate, amide, or ester functional groups were excluded as these would be unstable during the strong acid hydrolysis treatment required in the sample preparation steps; (3) ring compounds and organic salts were excluded; (4) unstable geminal diamines, which are believed to decompose into iminium species that can then hydrolyse further to aldehydes were also excluded [22]; and (5) hydrazines were excluded due to their apparently rare natural occurrence. Based on these criteria, the list of BMAA isomers was reduced to seven candidates (Fig. 1).

Structure of BMAA and selected isomeric compounds
One isomer in the list of candidates, DAB, is believed to be a hepatotoxic and neurotoxic non-protein amino acid that is found in many prokaryotic and eukaryotic organisms. It has already been widely investigated and can be distinguished from BMAA using recently developed methods based on LC and/or mass spectrometry [10,11,23]. Another BMAA isomer, N-(2-aminoethyl) glycine (AEG), has also been studied previously [24]. However, to date, β-amino-N-methyl-alanine (BAMA) has not been studied in the context of BMAA, even though it has the greatest potential among the selected candidates to yield the BMAA diagnostic product ion at m/z 258 when subjected to collision induced dissociation (CID). In this study, we analyzed the above-mentioned BMAA isomers, and showed that our improved LC-MS/MS platform based on AQC-derivatization is sufficient for distinguishing of BMAA from DAB, AEG, and BAMA as well as other potential isomers, enabling unambiguous identification of BMAA. Furthermore, high performance liquid chromatography (HPLC)- and ultra high-performance liquid chromatography (UHPLC-)-based methods were developed to extend the versatility of the application.
Experimental
Chemicals
β-N-methylamino-l-alanine hydrochloride (B107, Sigma-Aldrich, Germany) and L-2,4-diaminobutyric acid dihydrochloride (D8376, Sigma-Aldrich, Switzerland) were purchased. AEG was purchased from TCI (A1153, TCI, Japan), and BAMA was synthesized as described below.
Equipment
Mass spectrometry was carried out on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific, San Jose, USA) coupled with an Accela pump and Accela autosampler HPLC system (Thermo Fisher Scientific, San Jose, USA). The software package Xcalibur 2.1 was used to analyze the acquired data.
Synthesis of BAMA
The reaction strategy for the synthesis of BAMA is shown in Scheme 1. The synthetic method consisted of three steps:

The reaction scheme of BAMA synthesis
Firstly, to synthesize 5-(methylamino)-uracil, 5 g of 5-bromo-uracil was dissolved in a 15-ml solution of 33 % methylamine in ethanol contained in a large test tube which was then placed inside a steel bomb. The bomb was heated to 160 °C for 6 h, after which it was allowed to cool to room temperature. The precipitated crude product was filtered using a glass fritted funnel and then washed with water followed by acetone and diethylether. The desired product was obtained as white crystals with a yield of 64 %.
Secondly, to synthesize 5-(methylamino)-5, 6-dihydrouracil hydrochloride, 2.354 g of 5-(methylamino)-uracil was converted to its hydrochloric salt by treatment with 10 % HCl in methanol. Excess solvent and acid were removed in vacuo, and the salt was then suspended in 35-ml water inside a hydrogenation flask. A catalyst, comprising 5 % rhodium on alumina (306 mg) was added to the flask, and the mixture was hydrogenated at 50–60 psi for over 24 h, after which the catalyst was removed by filtering the mixture through a celite pad. Water was then removed azeotropically using toluene in vacuo. The resulting solid was sufficiently pure to be used directly in the subsequent reaction. The final yield of this step was 88 % [25].
Lastly, to synthesize BAMA, 2.375 g of 5-(methylamino)-5,6-dihydrouracil hydrochloride was dissolved in 30-ml 6 N HCl and then refluxed for 5 days in a flask. After cooling, the solvent was removed azeotropically using toluene in vacuo. To the resulting yellow-orange oil, boiling ethanol (ca. 15 ml) was added and water was added drop-wise until complete dissolution was achieved. Finally, acetone was added to the mixture until the product almost precipitated at reflux temperature. The flask was wrapped and left to cool overnight. The resulting white crystals were filtered and dried in vacuo. The filtrate was concentrated in vacuo, and the recrystallization procedure was repeated to recover further product. The second fraction was always slightly off-white, and this colour did not change upon repeated recrystallization. The final product was confirmed to be pure by NMR. The yield of this step was 58 % [25].
Preparation of biological samples
All samples analyzed in this study were kindly provided by Ulla Rasmussen (Department of Botany, Stockholm University). The field cyanobacteria, blue mussels and oysters were collected from the west coast of Sweden and stored at −20 °C until use.
The sample preparation was performed according to the method by Spacil et al. [1] with some modifications (Fig. 2). Briefly, the mussels and oysters were cut into thin strips and ground in liquid nitrogen using a mortar and pestle to get a solid–liquid mixture. Afterwards, 600 μl of this sample mixture were mixed with 600-μl 80 % methanol in water (v/v) and sonicated for nine cycles, 30 s each at 70 % intensity using an ultrasonicator (SONOPLUS, HP 2070, Bandelin electronic, Berlin, Germany). The cyanobacterial samples were mixed with 80 % methanol directly, then freezing/thawing in liquid nitrogen three times before ultrasonication. During the sonication, the sample tube was placed in an ice-water bath for 30 s in between two cycles to prevent protein degradation. Then the samples were centrifuged at 5,000×g for 3 min, and only the supernatants were used for further analysis.

The scheme of biological sample preparation for determining total BMAA
To determine protein concentration (RC/DC Protein Assay, Biorad, Sweden), 5 μl of sample supernatant was used in triplicates and the remaining material was dried in a speedvac (DNA 100 Speedvac, Savant) overnight at 55 °C. The dried samples were dissolved in 600 μL 6 M HCl and hydrolyzed at 110 °C for 20 h. The hydrolysates were filtered through the centrifugal filter (Ultrafree-MC, Millipore) at 12,200×g for 1 min, and the excess hydrochloric acid was evaporated using airflow at 55 °C in a heating block. The dried samples were reconstituted in 20 mM HCl solution to reach 12.5 mg/ml protein concentration. An aliquot (200 μl) of this solution containing 2.5 mg of protein equivalent was diluted by 800 μl 0.1 % formic acid in water (v/v) before solid phase extraction (SPE).
The SPE column (Isolute HCX-3, 100 mg, Sorbent AB, Sweden) was conditioned by 1 ml of methanol and equilibrated by 1 ml of 0.1 % (v/v) formic acid solution subsequently. After loading 1 ml of the sample, the column was washed by 1 ml of 0.1 % formic acid solution followed by 1 ml of 0.1 % formic acid in 25 % methanol (v/v). Finally, BMAA was eluted from the column by 1-ml 2 % ammonium hydroxide in methanol (v/v). The eluates were dried over night in a Speedvac at 50 °C. Finally, half of the material (i.e., 1.25 mg of protein equivalent) was reconstituted in 20-μl 20 mM HCl solution before derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) using Waters AccQ·Tag kit (WAT052880, Milford, MA, USA).
HPLC and UHPLC
HPLC separation was carried out with a Hypersil GOLD C18 column (100 × 2.1 mm, 3-μm particle size; Thermo Scientific, San Jose, CA) and a binary mobile phase (solvent A: 5 % acetonitrile in water with 0.3 % acetic acid and 0.005 % trifluoroacetic acid (TFA); solvent B: acetonitrile with 0.3 % acetic acid and 0.005 % TFA) delivered at a flow rate of 400 μl/min. The linear gradient elution program used was as follows: 0.0 min, 0 % B; 10.0 min, 10 % B; 15.0 min, 80 % B; 17.0 min, 80 % B; 18.0 min, 0 % B; and 25.0 min, 0 % B.
UHPLC separation was carried out with an Agilent Bonus RP Rapid Resolution High Throughput (RRHT) column (100 × 2.1 mm, 1.8 μm particle size; Agilent, USA) and a binary mobile phase (solvent A: 0.1 % formic acid in water; solvent B: 0.1 % formic acid in acetonitrile) delivered at a flow rate of 400 μl/min. The linear gradient elution program used was as follows: 0.0 min, 5 % B; 5.0 min, 15 % B; 7.5 min, 80 % B; 8.5 min, 80 % B; 9 min, 5 % B; and 13 min, 5 % B.
The HPLC or UHPLC chromatographic resolution evaluated between two adjacent peaks for BMAA, BAMA, DAB and AEG in the presence of different mobile-phase additives was calculated from the formula: R s = 2 (t RB – t RA )/(w B + w A ) where R s is the chromatographic resolution, t R is the retention time, and w is the width at the base of the chromatographic peak. B is the late-eluted and A is the early eluted analyte for two adjacent peaks.
Tandem mass spectrometry
AQC derivatives of BMAA were analyzed in positive ion detection mode using SRM scan type with the electrospray ionization (ESI) technique. The first quadrupole mass analyzer (Q1) was operated in high resolution mode with a full-width half maximum (FWHM) of 0.1 Da, whereas the second quadrupole mass analyzer (Q3) was operated at 0.7 FWHM resolution. For selective identification of BMAA, four SRM transitions were monitored (collision energies (CE) in eV are given in the brackets): 459.18 > 119.08 (CE = 20), 459.18 > 258.09 (CE = 25), 459.18 > 188.08 (CE = 35), 459.18 > 214.10 (CE = 35). The ion source parameters were optimized as below: spray voltage (5,000 V), vaporizer temperature (300 °C), capillary temperature (300 °C), sheath gas pressure (30 psi), ion sweep gas pressure (0 psi), auxiliary gas pressure (20 psi), S-lens (110), de-clustering voltage (0 V), and argon collision gas pressure (1.5 mTorr).
Results and discussion
BMAA and its isomers
The use of SRM with triple quadrupole mass spectrometers has proven to be the method-of-choice for biological analysis due to high sensitivity and selectivity in the presence of complex matrices. The first (Q1) and third (Q3) quadrupoles can operate either as ion transmission or mass filter devices, whereas the second (Q2) quadrupole functions as a collision cell. In an SRM experiment, both Q1 and Q3 function as mass filters by allowing only particular m/z precursor ions to pass through Q1 or product ions through Q3. Therefore the technique has very high specificity, enabling structure-based identification.
The unambiguous identification of AQC-derivatives of BMAA using the proposed LC-MS/MS method was ensured by a four-stage screening procedure, namely: (1) reversed-phase chromatographic separation of BMAA from its isomers DAB, BAMA and AEG; (2) isolation of ions with m/z 459.18 corresponding to AQC doubly labeled BMAA, at Q1; (3) monitoring one general (459.18 > 119.08) and one specific SRM transition (459.18 > 258.09 for BMAA/BAMA, 459.18 > 214.10 for AEG, and 459.18 > 188.08 for DAB) to obtain structural information for each compound; and 4) determination of the peak area ratios of two transitions (119.08/258.09 for BMAA/BAMA, 119.08/214.10 for AEG, and 119.08/188.08 for DAB) to enhance selectivity.
The TSQ Vantage triple-quadrupole mass analyzer has a capability of up to 0.1 FWHM mass resolution, which only allowed ions with a narrow mass range (0.1 Da) around the precursor ion to pass Q1 eliminating majority of interference by ions with m/z similar to BMAA derivates. Thus, increased selectivity can be achieved at the cost of decreased ion transmission. To evaluate loses in ion transmission, we tested 0.1, 0.2, 0.4, and 0.7 FWHM settings on Q1 using a BMAA spiked cyanobacterial sample (0.15 ng of derivatized BMAA was spiked in 2 mg (dry weight) of pre-treated cyanobacteria, Synechocystis sp. PCC6803). The peak area of 459.18 > 119.08 SRM signal from BMAA was used for comparison. For Q1 mass resolution setting at 0.7 to 0.4, 0.2, and 0.1 FWHM, the measured peak area of 459.18 > 119.08 SRM (n = 3) decreased by 1.12 %, 14.13 %, and 42.74 %, respectively. Considering signal interferences, data of equal quality were acquired for 0.2, 0.4, and 0.7 compared with 0.1 FWHM mass resolution setting in the sample we examined. However, we investigated 0.1 FWHM settings, because more complex biological samples, particularly samples from higher organisms may contain unknown interferences with precursor ion at m/z similar to BMAA. The high-resolution setting on Q1 helps to eliminate those putative interferences in Q1 which works as the first mass filter in a SRM procedure. For trace level of BMAA analysis in biological samples, 0.2 FWHM setting on Q1 is recommended without substantial loss of signal on a TSQ Vantage instrument. Loses in ion transmission by increasing mass resolution in Q1 must be determined experimentally when using other conventional quadrupole instrument. Together with the product ion at m/z 119.08, which corresponds to the protonated BMAA molecule as well as its isomers, another BMAA structure-specific product ion at m/z 258.09 was selected in Q3. The product ion at m/z 258.09 is generated by the cleavage of the C–N bond between the β carbon and the secondary amine nitrogen of BMAA, and therefore provides structure-specific information (Fig. 3a). This diagnostic ion can be used to distinguish BMAA from the isomers which do not generate product ion at m/z 258.09, e.g., DAB and AEG. The diagnostic product ions at m/z 188.08 for DAB and 214.10 for AEG were also found to be structure-specific and hence were used to differentiate them from BMAA (Fig. 3b, c). Combined with different LC properties of BMAA, DAB, and AEG, the MS/MS data allowed BMAA to be confidently distinguished from DAB and AEG as well as other potential isomers which do not yield m/z 258.09 product ions.

ESI-MS/MS product ion spectra of a BMAA (CE = 30 eV), b DAB (CE = 30 eV), c AEG (CE = 30 eV), and d BAMA (CE = 28 eV). The experiment was performed on a triple quadrupole mass spectrometer instrument API2000 (AB Sciex) using N2 as collision gas with gas pressure at 3e−5 Torr
In principle, BMAA isomers may co-elute with BMAA, again giving a product ion at m/z 258.09. According to our database search and the method selectivity criteria named above, BAMA was the only candidate that is stable in nature and can potentially yield the diagnostic product ion at m/z 258.09. For the remaining candidates, cleavage of the C-C bond to generate product ion at m/z 258.09 was not favorable under the CID conditions used in our method (Fig. 1). By BAMA synthesis and analysis of its AQC derivates using MS/MS, we confirmed that BAMA can yield the same product ions as BMAA, including the diagnostic ion at m/z 258.09 (Fig. 3a, d). Therefore, in an attempt to distinguish BMAA from BAMA, we evaluated the ratio of the two product ions at m/z 119.08 and 258.09. Based on the acquired MS/MS data, the yield of the product ion at m/z 258.09 for BMAA was higher than that of BAMA under the same CID conditions. The supposed fragmentation mechanisms and the structures of BMAA and BAMA product ions at m/z 258.09 are shown in the Fig. 4a, b, respectively. In both cases, the fragmentation mechanism is driven by proton transfer from α or β positions (of BMAA and BAMA, respectively) to the carbonyl oxygen of the respective ureido group. However, it is not possible to determine whether the adjacent charge is involved in the fragmentation or a charge-remote dissociation mechanism is at play. We have to consider both possibilities given the similar gas-phase basicities of the tertiary carbamate nitrogen and tertiary nitrogen in chinoline heterocycle. Further experiments are required to fully elucidate the gas phase chemistry. The practical result was that the ratio of 119.08/258.09 was found to be specific for BMAA and BAMA, and can be used to distinguish BMAA from BAMA as well as other potential isomers. The ratios for BMAA and BAMA measured in standard solutions with different concentrations are given in Table 1. For 0.1 ppm solution, the ratios were 4.4 and 18.4 for BMAA and BAMA, respectively. The consistency of the product ion ratios for BMAA and its isomers was investigated using standards solutions at three concentrations, corresponding to different 119.08 peak areas and representing levels expected in real samples. The calculated ratios showed an increasing trend from high to low concentration. The variability of repeated measurements (n = 5) of the 119.08/258.09 ratios was relatively higher at lower concentrations, probably due to distorted peak shapes, which would result in less accurate quantification. To determine the influence of sample matrix affects on the ratio, three different levels of BMAA, BAMA, and DAB standards were spiked into 1.25 mg protein equivalent of cyanobacterial matrix (Synechococcus sp.). AEG was not used because it co-eluted with an interference present in the sample matrix. The results showed that the SRM product ion ratios correlate well with those in standard solutions, if the analytes were separated on the LC column (Table 1). Based on these results, it can be concluded that for the identification of BMAA in biological samples, the ratio of 119.08/258.09 should be consistent with the ratio measured in BMAA standard solutions at similar concentrations, within an error of ±10 %. However, two situations may cause the ratio to be significantly higher than the standard reference solutions at similar concentration. Firstly, an isomer, such as BAMA may give a product ion at m/z 258.09, which consequently shifts the ratio of 119/258. Alternatively, an isomer, such as DAB or AEG, may co-elute with BMAA, which would give a product ion peak at m/z 119.08 but not 258.09.

Suggested fragmentation pathways and gas phase ion structures of a) BMAA; and b) BAMA-AQC derivates
Separation using HPLC and UHPLC
The HPLC separation was optimized using different mobile phase additives in a binary eluent system (eluent A, 5 % (v/v) acetonitrile in water and eluent B, acetonitrile) with the linear gradients condition and flow rates described in the “Experimental” section. Formic acid (0.1 %, v/v) in both eluents was tested initially, but BMAA and AEG were found to co-elute completely (Fig. 5a). Acetic acid (0.3 %, v/v) was therefore chosen to alter the chromatographic selectivity of the LC separation. Under this condition, the BMAA peak was partly separated from that for AEG, and the MS/MS detection sensitivity was improved. However, the chromatographic peak shapes were distorted (Fig. 5b). Therefore, TFA at a concentration of 0.005 % (v/v) was added to the mobile phase in order to improve the peak shape and thus separation efficiency (Fig. 5c), despite TFA is known to cause strong ion suppression during ESI particularly for basic compounds [26]. Pentadecafluorooctanoic acid (PDFOA) was also tested as an alternative to TFA but with less ion suppression potential [27]. Using PDFOA, the ion suppression effect was reduced, but the signal-to-noise ratio was not significantly improved and excessive peak broadening was also apparent. Thus PDFOA was not employed further. The mobile phase with 0.3 % acetic acid resulted in the method with the lowest detection limit, while the mobile phase with 0.3 % acetic acid and 0.005 % TFA gave the highest performance suitable to separate all studied BMAA isomers. The repeatability of the retention time (n = 5) for each compound is shown in Table 1. To account for the method sensitivity when using 0.3 % acetic acid as mobile phase additive, 2 mg (dry weight) of pre-treated cyanobacteria (Synechocystis sp. PCC6803) was spiked with 0.2 ng BMAA before derivatization. The signal-to-noise ratio of 22 was measured for 459.18 > 258.09 SRM transition from BMAA, thus detection limit in cyanobacteria is below 0.1 μg/g dry weight, assuming 100 % recovery during sample preparation.

LC-MS/MS chromatograms showing the effects of different mobile-phase additives on BMAA, BAMA, AEG, and DAB analysis by HPLC—a 0.1 % formic acid, b 0.3 % acetic acid, c 0.3 % acetic acid + 0.005 % TFA and by UHPLC—d 0.1 % formic acid
The UHPLC separation was performed using an Agilent Bonus RP RRHT column (100 × 2.1 mm, 1.8 μm particle size). This column has a polar-embedded C14 stationary phase which gives an alternative selectivity compared with the C18 column and a stable performance when using highly aqueous mobile phases. Indeed, the elution order of BMAA and AEG was reversed on this column compared with the C18 stationary phase (as used in the HPLC method described above), which allows for identification of some interfering isomers. We also achieved a relatively high throughput separation of BMAA, and its isomers within 13 min, including column wash and equilibration steps (Fig. 5d). The HPLC and UHPLC chromatographic resolution between BMAA, and its three isomers at different conditions was calculated and listed in Table 2.
The biological samples
Initially, we examined laboratory-cultured cyanobacterial samples by HPLC-MS/MS with 0.1 % formic acid in the mobile phase. Under these conditions, we often observed the m/z 119.08 SRM ion trace peak occurred at an equivalent retention time to the BMAA standard. However, the diagnostic SRM ion trace peak at m/z 258.09 detected for the samples was either missing or significantly less intense than in the BMAA standard solutions while the peak areas of 119.08 SRM ion trace were similar for both the samples and standard solutions. Consequently, the resulting 119.08/258.09 ratio for the biological samples was inconsistent with that obtained for the BMAA standard. When the cyanobacterial samples were spiked with the BMAA standard, the SRM chromatograms revealed that the BMAA from the standard co-eluted with the compound giving the ions at m/z 119.08 in the cyanobacterial samples and suggested the presence of an impurity. The absence of the 258.09 peak may be due to: (1) insufficient concentration of BMAA in the sample, since the intensity of the 258.09 peak is about 4.5 times lower than the intensity of the 119.08 peak. Therefore, it is possible that the 119.08 SRM ion trace is detected, but the intensity of the 258.09 SRM ion trace was below the detection limit or (2) the presence of a BMAA isomer, which exclusively gives an SRM ion trace at 119.08, but not 258.09 (e.g., AEG). The elevated 119.08/258.09 ratio may arise because: (1) BMAA co-elutes with an interfering compound which does not generate a product ion at m/z 258.09 or (2) a BMAA isomer is present, which generates product ions at both m/z 119.08 and 258.09, and elutes with a similar retention time as BMAA, but has a higher 119.08/258.09 product ion ratio.
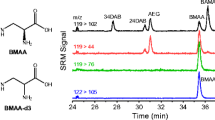
We further analyzed some field samples of cyanobacteria, blue mussel and oyster from a related study using the HPLC-MS/MS method with 0.3 % acetic acid as mobile-phase additive. We often observed a peak of the 459.18 > 119.08 SRM transition by an interference that eluted close to the expected AEG retention time. However, AEG qualifier peak at m/z 214.10 was either missing or at lower intensity than that detected for AEG standard. We were not able to assign a structure to this interference based on current data. As an example, we spiked a BMAA-negative laboratory-cultured (Synechococcus sp.) and field-collected cyanobacteria (Fig. 6a, b) from the west coast of Sweden with a BMAA standard addition. (The injected amount of BMAA was 10 pg spiked in 250 μg protein equivalent of cyanobacteria.) The results showed the interference peak appeared in the immediate vicinity of the BMAA peak with the chromatographic resolution of 0.83 and 0.93 for laboratory-cultured and field-collected cyanobacteria, respectively. Similar results were obtained with blue mussel (Mytilus edulis) and oyster (Ostrea edulis) samples from west coast of Sweden, which contained BMAA (Fig. 6c, d). The chromatographic resolutions between the interference and BMAA were 0.98 and 0.85 for blue mussel and oyster, respectively. The comprehensive identification of the interfering compound is unfortunately beyond the scope of this study. Interestingly, we also detected BAMA in these blue mussel and oyster samples. BAMA was well separated from BMAA by LC retention time and the ratio of 119.08/258.09 was higher for BAMA (ca. 19.5) compared with BMAA (4.3), allowing these two compounds to be readily distinguished using the proposed SRM detection method. Nevertheless, the presence of BAMA in real samples may give false positives because several of the product ions generated from BAMA are identical to BMAA, including the diagnostic product ion at m/z 258.09. Additionally, the presence of the interference in most of real samples has to be considered as this interference may complicate BMAA identification due to very similar retention properties and generation of product ion at m/z 119.08. Therefore, errors in BMAA identification are possible if the methods of separation and detection are not optimized carefully.

LC-MS/MS chromatograms of biological samples a BMAA-negative laboratory-cultured cyanobacterial sample spiked with BMAA standard, b BMAA-negative field cyanobacterial sample spiked with BMAA standard, c BMAA-positive field blue mussel field sample, d BMAA-positive field oyster field sample. Identification based on retention times and characteristic SRM ion traces was confirmed comparison with a BMAA standard solution. The retention time shift observed in samples (a) and (b) versus (c) and (d) was attributed to using a new batch of HPLC column
Several studies have reported interfering compounds or isomers co-eluting with BMAA. DAB was the first BMAA isomer found in a cyanobacteria sample (Calothrix PCC7103) which has similar retention characteristics to BMAA [11]. Another research group discovered several compounds in animal tissue samples which eluted in close proximity to BMAA, including a compound that completely co-eluted with BMAA in the first but not the second dimension of gas chromatography data [14]. Our study here complements and further extends the previous reports in common effort of unequivocal differentiation of BMAA from its isomers using either HPLC or UHPLC separation coupled with MS/MS detection since the unknown interfering compound(s) eluting slightly later than BMAA were often observed in the cyanobacterial samples.
Conclusions
This paper presents a reliable protocol for BMAA identification while addressing issues concerning all known BMAA isomers, by using either HPLC or UHPLC coupled to MS/MS. The presence of a BMAA isomer, BAMA, is reported for the first time in natural samples. The frequently observed interfering compound(s) in biological samples can be well separated by our UHPLC- or partially separated by our HPLC method, and further distinguished unambiguously from BMAA by MS/MS procedure, although it is not possible to assign a structure to this interference based on current data. Nevertheless, the analytical approach we present here proves to be a robust method for distinguishing BMAA from complex biological matrices and will contribute to resolve the controversial findings reported to date in the BMAA research field. This is particularly important because the toxicity pathway of BMAA has not yet been clearly established. Hence, the availability of a reliable analytical method is essential for the correct identification of BMAA and to prevent the misinterpretation of data.
References
Spacil Z, Eriksson J, Jonasson S et al (2010) Analytical protocol for identification of BMAA and DAB in biological samples. Analyst 135:127
Cox P, Banack S, Murch S (2003) Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc Natl Acad Sci USA 100:13380
Cox PA, Banack SA, Murch SJ et al (2005) Diverse taxa of cyanobacteria produce beta-N-methylamino-l-alanine, a neurotoxic amino acid. Proc Natl Acad Sci USA 102:5074
Banack SA, Johnson HE, Cheng R et al (2007) Production of the neurotoxin BMAA by a marine cyanobacterium. Mar Drugs 5:180
Johnson HE, King SR, Banack SA et al (2008) Cyanobacteria (Nostoc commune) used as a dietary item in the Peruvian highlands produce the neurotoxic amino acid BMAA. J Ethnopharmacol 118:159
Esterhuizen M, Downing T (2008) Beta-N-methylamino-l-alanine (BMAA) in novel South African cyanobacterial isolates. Ecotoxicol Environ Saf 71:309
Metcalf JS, Banack SA, Lindsay J et al (2008) Co-occurrence of beta-N-methylamino-l-alanine, a neurotoxic amino acid with other cyanobacterial toxins in British waterbodies, 1990–2004. Environ Microbiol 10:702
Eriksson J, Jonasson S, Papaefthimiou D et al (2009) Improving derivatization efficiency of BMAA utilizing AccQ-Tag in a complex cyanobacterial matrix. Amino Acids 36:43
Bidigare RR, Christensen SJ, Wilde SB et al (2009) Cyanobacteria and BMAA: possible linkage with avian vacuolar myelinopathy (AVM) in the south-eastern United States. Amyotroph Lateral Scler 10(Suppl 2):71
Faassen EJ, Gillissen F, Zweers HAJ et al (2009) Determination of the neurotoxins BMAA (beta-N-methylamino-l-alanine) and DAB (alpha-, gamma-diaminobutyric acid) by LC-MSMS in Dutch urban waters with cyanobacterial blooms. Amyotrophic Lateral Scler 10:79
Rosen J, Hellenaes K (2008) Determination of the neurotoxin BMAA (β-N-methylamino-l-alanine) in cycad seed and cyanobacteria by LC-MS/MS (liquid chromatography tandem mass spectrometry). Analyst (Cambridge, U K) 133:1785
Kisby GE, Roy DN, Spencer PS (1988) Determination of β-N-methylamino-l-alanine (BMAA) in plant (Cycas circinalis L.) and animal tissue by precolumn derivatization with 9-fluorenylmethyl chloroformate (FMOC) and reversed-phase high-performance liquid chromatography. J Neurosci Methods 26:45
Snyder LR, Cruz-Aguado R, Sadilek M et al (2009) Parkinson-dementia complex and development of a new stable isotope dilution assay for BMAA detection in tissue. Toxicol Appl Pharmacol 240:180
Snyder LR, Hoggard JC, Montine TJ et al (2010) Development and application of a comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry method for the analysis of l-beta-methylamino-alanine in human tissue. J Chromatogr A 1217:4639
Marler TE, Snyder LR, Shaw CA (2010) Cycas micronesica (Cycadales) plants devoid of endophytic cyanobacteria increase in β-methylamino-l-alanine. Toxicon 56:563
Caller TA, Doolin JW, Haney JF et al (2009) A cluster of amyotrophic lateral sclerosis in New Hampshire: a possible role for toxic cyanobacteria blooms. Amyotrophic Lateral Scler 10:101
Cox PA, Richer R, Metcalf JS et al (2009) Cyanobacteria and BMAA exposure from desert dust: a possible link to sporadic ALS among Gulf War veterans. Amyotrophic Lateral Scler 10:109
Craighead D, Metcalf JS, Banack SA et al (2009) Presence of the neurotoxic amino acids beta-N-methylamino-l-alanine (BMAA) and 2,4-diamino-butyric acid (DAB) in shallow springs from the Gobi Desert. Amyotrophic Lateral Scler 10:96
Roney BR, Li R, Banack SA et al (2009) Consumption of fa cai Nostoc soup: a potential for BMAA exposure from Nostoc cyanobacteria in China? Amyotrophic Lateral Scler 10:44
Jonasson S, Eriksson J, Berntzon L et al (2010) Transfer of a cyanobacterial neurotoxin within a temperate aquatic ecosystem suggests pathways for human exposure. Proc Natl Acad Sci USA 107:9252
Kruger T, Monch B, Oppenhauser S et al (2010) LC-MS/MS determination of the isomeric neurotoxins BMAA (beta-N-methylamino-l-alanine) and DAB (2,4-diaminobutyric acid) in cyanobacteria and seeds of Cycas revoluta and Lathyrus latifolius. Toxicon 55:547
Moad G, Benkovic SJ (1978) On the mechanism of decomposition of geminal diamines. J Am Chem Soc 100:5495
Banack S, Downing T, Spacil Z et al (2010) Distinguishing the cyanobacterial neurotoxin beta-N-methylamino-l-alanine (BMAA) from its structural isomer 2,4-diaminobutyric acid (2,4-DAB). Toxicon 56:868
Banack SA, Metcalf JS, Spacil Z et al (2011) Distinguishing the cyanobacterial neurotoxin β-N-methylamino-l-alanine (BMAA) from other diamino acids. Toxicon 57:730
Dietrich RF, Sakurai T, Kenyon GL (1979) Substituted beta.-alanines. Preparation of.alpha.-substituted.beta.-alanine derivatives from 5-substituted uracils and dihydrouracils. J Org Chem 44:1894
Apffel A, Fischer S, Goldberg G et al (1995) Enhanced sensitivity for peptide mapping with electrospray liquid chromatography–mass spectrometry in the presence of signal suppression due to trifluoroacetic acid-containing mobile phases. J Chromatogr A 712:177
Aruda WO, Aruda KO (2009) Review of volatile perfluorocarboxylic acids as ion-pair reagents in LC, part II. LCGC North Am 27:916–928
Acknowledgments
We thank Ulla Rasmussen (Department of Botany, Stockholm University) for kindly providing the biological samples in this study. We also thank Frank Turecek (University of Washington, Seattle, WA 98195, USA) for his suggestions concerning the gas-phase chemistry and Peter B. Wyatt (School of Biological and Chemical Sciences at Queen Mary University of London) for helpful discussions.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Jiang, L., Aigret, B., De Borggraeve, W.M. et al. Selective LC-MS/MS method for the identification of BMAA from its isomers in biological samples. Anal Bioanal Chem 403, 1719–1730 (2012). https://doi.org/10.1007/s00216-012-5966-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-5966-y