
Abstract
Glycoproteins secreted or expressed on the cell surface at specific pathophysiological stages are well-recognized disease biomarkers and therapeutic targets. While mapping of specific glycan structures can be performed at the level of released glycans, site-specific glycosylation and identification of specific protein carriers can only be determined by analysis of glycopeptides. A key enabling step in mass spectrometry (MS)-based glycoproteomics is the ability to selectively or non-selectively enrich for the glycopeptides from a total pool of a digested proteome for MS analysis since the highly heterogeneous glycopeptides are usually present at low abundance and ionize poorly compared with non-glycosylated peptides. Among the most common approaches for non-destructive and non-glycan-selective glycopeptide enrichment are strategies based on various forms of hydrophilic interaction liquid chromatography (HILIC). We present here a variation of this method using amine-derivatized Fe3O4 nanoparticles, in concert with in situ peptide N-glycosidase F digestion for direct matrix-assisted laser desorption/ionization–mass spectrometry analysis of N-glycosylation sites and the released glycans. Conditions were also optimized for efficient elution of the enriched glycopeptides from the nanoparticles for on-line nanoflow liquid chromatography–MS/MS analysis. Successful applications to single glycoproteins as well as total proteomic mixtures derived from biological fluids established the unrivaled practical versatility of this method, with enrichment efficiency comparable to other HILIC-based methods.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycosylation is one of the most common post-translational protein modifications in nature and is increasingly found to affect the physicochemical properties and biological activities of proteins in a site-specific manner. Almost without exception, each glycosylation site is endowed with a defined range of glycan heterogeneity that mediates functions as diverse as protein maturation and turnover, cell adhesion and trafficking, receptor binding, and activation [1, 2]. Aberrant glycosylation leading to tumor-associated carbohydrate antigens is a well-recognized trademark of cancer cells [3]. This and several well-documented developmental changes in the global glycosylation profiles have, in fact, driven most of the current glycomic analyses, which focus on identifying the implicated glycans without addressing the protein carriers [4, 5]. However, it is now commonly appreciated that sensitive detection of protein-specific glycosylation changes associated with disease or physiological activation states is essential not only for developing biomarkers of better selectivity [6] but also underlies most basic glycobiology studies.
Despite recent advances in mass spectrometry (MS)-based proteomics, protein glycosylation analysis remains a formidable technical challenge, either on a global scale or for isolated individual glycoproteins of interest [4, 6, 7]. Among the limiting factors are their higher molecular weight, lower abundance, and ionization efficiency relative to non-glycosylated peptides, such that the individual glycopeptide ion signals are often suppressed in MS analysis of unfractionated peptide/glycopeptide mixtures. Thus, an ability to indiscriminately enrich for the glycopeptides from a proteolytic digest of a proteome or an isolated glycoproteome is a pre-requisite to MS-based glycoproteomics, which aims to globally map the site-specific distribution of individual glycans from a specified glycomic repertoire onto their respective protein carriers. Likewise, to define an unknown protein glycosylation profile or to monitor for the glycosylation fidelity of a wide range of recombinant glycoprotein therapeutics including the biosimilars, a non-selective enrichment method of glycopeptides is needed lest a particular glycoform is under-represented. This requirement differs from the enrichment strategy in which glycopeptides carrying a pre-determined, specific glycotope are targeted for identification.
For selective enrichment, one or multi-dimensional lectin affinity capture in various formats [8–10] are the most commonly used approaches and have been very successful for comparative analysis of the targeted sub-glycoproteomes carrying particular glycan features. Alternatively, the negatively charged, sialic acid-containing glycopeptides can be efficiently retained by titanium dioxide and eluted under specific buffer conditions [11]. On the other hand, to non-selectively capture a full range of glycopeptides irrespective of the attached glycan structures, the working principle has always been relying on the fact that glycopeptides are, in general, not only larger in size but also more hydrophilic than non-glycosylated peptides. Glycopeptides can thus be significantly enriched by size exclusion chromatography [12] or, more commonly, via various forms of hydrophilic interaction liquid chromatography (HILIC) [7], which includes carbohydrate gel matrices, such as cellulose [13], maltose [14], or Sepharose [15], amide-based [16], and zwitterionic type (ZIC-HILIC) [17–19] stationary phases.
A different approach for general glycopeptide enrichment targets the common glycan moieties. The affinity of porous graphitic carbon stationary phase for oligosaccharides has been used to isolate and separate glycopeptides provided the carrier peptides are no more than a few amino acids. It is thus best suited for extensive proteolytic digests but may preclude confident protein identification in glycoproteomic applications [20–22]. Hydrazide functionalized beads can trap glycoproteins and glycopeptides by covalent binding after periodate oxidation of the adjacent diols of saccharides [23], but this method suffers from the loss of the glycans in the process of releasing the captured glycopeptides via de-N-glycosylation. Overall, the HILIC approach, either by solid phase extraction or by direct liquid chromatography (LC)-MS/MS coupling, appears to be the most convenient and general enrichment method without a need for initial chemical derivation and/or more cleaning-up steps that may compromise yield.
Taking this a step further, functionalized magnetic beads or nanoparticles have recently gained popularity in proteomics research with respect to enabling a convenient and rapid detection by MS [24]. Coupling of di-boronic acid or lectins such as concanavalin A and wheat germ agglutinin was shown to selectively concentrate glycoproteins or glycopeptides [25–27], but the efficiency against more complex mixtures has only been reported for few biological samples [26, 27]. The magnetic nanoparticles are, however, very attractive as enrichment beads since they can additionally be spotted directly for matrix-assisted laser desorption/ionization–mass spectrometry (MALDI-MS) detection along with many possible on-bead enzymatic or chemical treatments. Retained analytes can also be eluted off for LC-MS/MS applications after extensive washes. It thus represents a good vehicle to implement HILIC for a more versatile, efficient, and non-selective enrichment of all glycopeptides that will fulfill both the requirements in enabling shotgun glycoproteomics and rapid single protein glycosylation analysis.
We demonstrate in this work an integrated analytical approach for glycosylation analysis facilitated by the enrichment of glycopeptides based on amine-derivatized Fe3O4 nanoparticles. Both proteolytic peptides and glycopeptides can be trapped through hydrophilic and electrostatic interactions but glycopeptides were retained more tightly, most likely through the additional interactions contributed by glycan moiety. After washing off the non-glycopeptides by adjusting the pH value, glycopeptide-bound nanoparticles could be spotted onto MALDI target plate and directly desorbed for MS and MS/MS analysis, which is particularly useful when handling low femtomole samples. Additional in situ deglycosylation by peptide N-glycosidase F (PNGase F) would allow ready determination of N-glycosylation sites with minimal sample loss. Alternatively, glycopeptides can be eluted off the nanoparticles for on-line LC-electronspray ionization–MS/MS analysis, which is useful for more complex biological samples. The simplicity and versatile utilities of this non-selective glycopeptide enrichment method should make it a useful enabling tool for both single glycoprotein and glycoproteomic analyses.
Materials and methods
Materials
Iron (II and III) oxide nanopowder (Fe3O4, 20–30 nm), 3-aminopropyl trimethoxysilane, bovine fetuin, asialofetuin, α-cyano-4-hydroxy-cinnamic acid (CHCA), 2,5-dihydroxybenzoic acid (DHB), phosphoric acid, and ammonium bicarbonate were obtained from Sigma-Aldrich (St. Louis, MO); PNGase F from Roche Diagnostics (Mannheim, Germany); sequencing grade, modified trypsin from Promega (Madison, WI); and formic acid, ammonia solution (NH4OH), acetonitrile, and ethanol from Merck (Darmstadt, Germany). Recombinant erythropoietin (EPO) from BHK21 cells was purchased from Advanced Gene Technology Corp (Taiwan). Uterine luminal fluid (ULF) was collected from 3-week-old female mice pretreated with diethylstilbestrol as described previously [28]. Mouse serum was collected from BALB/cJ mice (8 weeks, 25 g) and albumin was removed as described [29].
Preparation of amine-based magnetic nanoparticles
Fe3O4 nanoparticles were coated with amine as described [30, 31]. In brief, 10 mg of Fe3O4 nanoparticles were added to a freshly prepared 2% (v/v) solution of 3-aminopropyl trimethoxysilane (20 μl in 970 μl ethanol and 10 μl water), followed by sonication and then vigorous stirring at 45 °C for 6 h. Upon completion of the reaction, the nanoparticles were absorbed onto the wall of the tube by positioning a magnet, which allowed the remaining solution to be readily removed by pipet. The isolated nanoparticles were then sequentially washed with methanol, distilled water and acetonitrile with each washing step repeated twice in order to remove any unreacted impurities. Finally, the derivatized nanoparticles were resuspended in 1 ml of 95% acetonitrile/0.1% formic acid and stored at room temperature.
Proteolytic digestions
Stock solutions of proteins (approximately 10 μg) were reduced with 5 mM dithiothreitol at 37 °C for 1 h, alkylated with 10 mM iodoacetamide in 25 mM ammonium bicarbonate buffer at 37 °C for 1 h in the dark at room temperature, and then treated overnight with sequencing grade trypsin at an enzyme-to-substrate ratio of 1:50 at 37 °C as described previously [32]. The digested products were then diluted with 0.1% formic acid to proper concentration for further experiments.
Enrichment of glycopeptides by functionalized nanoparticles
In the present study, 1 μl of peptide digest solution (about 1 pmol for standard protein and 10 μg for complex protein mixtures) was added to 10 μl of the nanoparticles solution (10 mg/ml in 95% acetonitrile/0.1% formic acid) and the mixture was sonicated for 10 s to thoroughly suspend the nanoparticles. After 1 min incubation, the nanoparticles were absorbed by magnet onto the wall and the solution was removed. The isolated nanoparticles were washed with 95% acetonitrile/0.1% formic acid solution (2 × 10 μl) to remove salt and other impurities. To remove non-specifically bound non-glycosylated peptides, the nanoparticles were sequentially washed with 80% acetonitrile/1% H3PO4 and 80% acetonitrile/1% NH4OH (2 × 10 μl for each). For direct MALDI-MS analysis, 10 μl of CHCA solution (5 mg/ml in 75% acetonitrile/0.1% formic acid) was then added to re-suspend the nanoparticles and 1 μl of the mixture (about 100 fmol of single protein digest) was spotted directly onto MALDI target plate. Alternatively, the bound glycopeptides were eluted off the nanoparticles by repeated sonication in 10 μl of 50% acetonitrile/2% NH4OH for five times and collected solutions from each treatment were pooled, dried down by SpeedVac, and re-dissolved in 75% acetonitrile/0.1% formic acid for MALDI-MS analysis or 0.1% formic acid for LC-MS/MS analysis. For complex mixtures such as the ULF and mouse sera, one fifth of the recovered samples from the initial 10 μg used were taken for the LC-MS/MS based glycopeptide analysis.
Enrichment of glycopeptides by Sepharose
The hydrophilic affinity enrichment of glycopeptides by Sepharose was carried out based on the method described by Wada et al. [15]. About 10 μg of the proteolytic digest was added to a microcentrifuge tube containing 20 μl of Sepharose CL-4B resin (Amersham Bioscience, Piscataway, NJ) in 500 μl of organic solvent mixture containing 1-butanol/ethanol/water (4:1:1 by volume). The resulting mixture was gently mixed for 45 min, and then centrifuged for 10 min. The supernatant was removed, and the pellet was washed twice with the same organic solvent. The glycopeptides were extracted by incubating the pellet in 500 μl ethanol/water (1:1 by volume) solution for 30 min with shaking. The mixture was then centrifuged, and the supernatant was transferred to another microcentrifuge tube and collected. The same extraction procedure was repeated two times. Finally, the combined supernatant was vacuum dried.
Enzymatic de-N-glycosylation
For in situ de-N-glycosylation, glycopeptide bound nanoparticles were re-suspended in 25-mM ammonium bicarbonate, pH 8.5 and treated with 1 mU of PNGase F at 37 °C overnight. Subsequently, 1 μl of the de-N-glycosylated mixture was mixed with either 4 μl of CHCA solution or 10 mg/ml DHB solution in acetonitrile and 1-μl aliquots were spotted directly onto target plate for N-glycosylation site determination or glycan mapping, respectively, by MALDI-MS and MS/MS analysis. Glycopeptides eluted from nanoparticles or enriched by Sepharose resin were re-suspended in 25-mM ammonium bicarbonate, pH 8.5 and treated overnight with 1 mU of PNGase F at 37 °C. De-N-glycosylated peptides were then separated from the released N-glycans by using a C18 Zip-Tip (Millipore, Bedford, MA). Bound peptides were eluted off the Zip-Tip with 75% acetonitrile/0.1% formic acid.
MALDI-MS and MS/MS analysis of peptides
Glycopeptide detection in MALDI-MS linear mode and MS/MS sequencing of peptides in reflectron mode were performed on 4,700 Proteomics Analyzer mass spectrometer (Applied Biosystems, Framingham, MA) equipped with an Nd:YAG laser (355 nm wavelength, <500-ps pulse, and 200 Hz repetition rate in both MS and MS/MS modes). 1,000 and 5,000 shots were accumulated in positive ion mode MS and MS/MS, respectively. For collision-induced dissociation (CID) MS/MS operation, the indicated collision cell pressure was increased from 3.0 × 108 Torr (no collision gas) to 5.0 × 107 Torr, with the potential difference between the source acceleration voltage and the collision cell set at 1 kV. The resolution of timed ion selector for precursor ion was set at 200. All MALDI-MS/MS data were manually acquired and assigned to verify the sequences for N-glycosylation sites.
MALDI-MS glycan profiling
N-glycans released from nanoparticle-bound glycopeptides by PNGase F were directly profiled by MALDI-MS as described above, or after permethylation by the NaOH/dimethyl sulfoxide slurry method [33]. For permethylation, the de-N-glycosylated mixtures containing the NH2-nanoparticles were dried down before re-suspending the whole lot in the NaOH/dimethyl sulfoxide slurry, followed by addition of methyl iodide. The permethyl derivatives were extracted into chloroform, washed repeatedly with water, dried down, and then mixed 1:1 with DHB matrix solution for application to a MALDI target plate. Data acquisition was performed manually on the 4,700 Proteomics Analyzer operated in the reflectron mode.
Shotgun analysis of glycopeptides and de-N-glycosylated peptides by LC-MS/MS
For shotgun analysis of glycopeptides, online nanoLC-nanoESI-MS survey scan and fully automated data-dependent acquisition of CID MS/MS were performed on the Micromass Q-TOF Ultima API mass spectrometer under the full software control of MassLynx™ 4.1; 1 s survey scans were acquired over the mass range m/z 400–2,000 and a maximum of three concurrent MS/MS acquisitions could be triggered for 2+, 3+, and 4+ charged precursors detected at an intensity above the predefined threshold. Each MS/MS acquisition was completed and the instrument switched back to MS survey scan mode when the precursor intensity fell below a predefined threshold or after a maximum of 6 s acquisition. Glycopeptides were manually identified by the presence of glycan-specific oxonium ion fragments, m/z 204.08 for N-acetylhexosamine (HexNAc), m/z 292.10 for N-acetylneuraminic acid (NeuAc), and m/z 366.14 for hexose-N-acetylhexosamine (Hex-HexNAc) in the MS/MS spectra.
For shotgun analysis of de-N-glycosylated peptides, nanoLC–nanoESI-MS/MS analysis was performed on a nanoAcquity system (Waters, Milford, MA) connected to an LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Electron, Bremen, Germany) equipped with a PicoView nanospray interface (New Objective, Woburn, MA). Peptide mixtures were loaded onto a 75 μm ID, 25 cm length C18 BEH column (Waters, Milford, MA) packed with 1.7 μm particles with a pore size of 130 Å and were separated in 30 min using a gradient of 5% to 50% solvent B (acetonitrile with 0.1% formic acid) at a flow rate of 300 nl/min and a column temperature of 35 °C. Solvent A was 0.1% formic acid in water. The mass spectrometer was operated in the data-dependant mode. Briefly, survey full scan MS spectra were acquired in the Orbitrap (m/z 350–1,600) with the resolution set to 60,000 at m/z 400 and automatic gain control (AGC) target at 106. The ten most intense ions were sequentially isolated for CID MS/MS fragmentation and detection in the linear ion trap (AGC target at 7,000) with previously selected ions dynamically excluded for 90 s. Ions with singly and unrecognized charge state were also excluded. Raw data were transformed to msm-files using the software RAW2MSM (versions 1.10) [34] and searched against IPI mouse database (3.68), using an in house Mascot Daemon 2.3 server. Search criteria used were trypsin digestion, variable modifications set as carbamidomethyl (C), oxidation (M), and deamidation (NQ), allowing up to one missed cleavage, mass accuracy of 10 ppm on the parent ion, and 0.60 Da on the fragment ions. All significant protein hits from Mascot (p < 0.01) with ion scores of >34 were reported with an overall false discovery rate at <2.7%.
Results and discussion
Enrichment of glycopeptides by NH2-functionalized nanoparticles
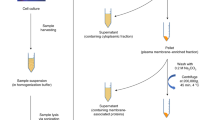
It was reasoned that although most tryptic peptides could be trapped on NH2-functionalized magnetic nanoparticles via hydrophilic and electrostatic interactions, glycopeptides would be bound much stronger than non-glycosylated ones due to the additional contribution of hydrophilic glycan moiety. Sialylated and/or sulfated glycopeptides may be adsorbed even more tightly via extra electrostatic interaction between the negatively charged carboxyl group on the sialic acid or sulfate and the amino group on the functionalized nanoparticles. Efficient glycopeptide enrichment could therefore be realized by developing proper binding, washing and sequential elution conditions. The conceptualized experimental workflow from glycopeptide enrichment to sequencing of both peptides and glycans as facilitated by the NH2-functionalized nanoparticles is schematically illustrated in Fig. 1, which includes the key steps developed.

A schematic workflow of the developed NH2-nanoparticles glycopeptide enrichment strategy, showing the sequential steps leading to either direct MALDI-MS detection, or further elution for LC-MS/MS analysis
Tested first against the sialylated glycopeptides derived from a tryptic digest of bovine fetuin, it was found that pH value was the most critical factor to selectively remove non-glycopeptides while retaining glycopeptides. The washing conditions were initially optimized by different concentrations of acetonitrile. 95% Acetonitrile/0.1% formic acid was found to remove most nonspecific binding peptides but retaining the glycopeptides and some charged peptides through electrostatic interaction. Both acidic conditions with 1–5% phosphoric acid/80% acetonitrile and basic conditions with 1–5% ammonia/80% acetonitrile were then tested. Finally, 1% phosphoric acid/80% acetonitrile at pH 1.2 and 1% ammonia/80% acetonitrile at pH 11.2 were found to most efficiently remove non-specific binding peptides whereas higher concentrations of ammonia led to partial elution of glycopeptides. Without any enrichment step, only weak signals corresponding to high molecular weight glycopeptides (m/z 3,000–7,000) could be detected by MALDI-MS analysis (Fig. 2A). After differential washing with high percentage of acetonitrile content coupled with 1% phosphoric acid and ammonia, glycopeptides corresponding to all three glycosylation sites of fetuin [35] could be clearly detected (Table 1). The MS spectrum in positive ion linear mode revealed a series of molecular ion signals differed by 291 Da, which was indicative of glycopeptides with different degree of sialylation (Fig. 2B). Any loss during the wash steps was negligible as no apparent glycopeptide signal could be detected by MALDI-MS for the wash fractions collected (Electronic supplementary material (ESM) Fig. S1b and S1c).

MALDI-MS spectra of bovine fetuin and asialofetuin tryptic peptides acquired in positive ion linear mode. Before enrichment, 100 fmol of both the fetuin (A) and asialofetuin (C) afforded mostly the non-glycosylated tryptic peptides, labeled as T x , where x denotes the order of the predicted tryptic peptides in series as counted from the N terminus. The non-labeled signals could be assigned as glycopeptides (Table 1), which became more prominent in the corresponding spectra afforded by the fetuin (C) and asialofetuin (D) tryptic digest samples extracted with NH2-functionalized Fe3O4 nanoparticles. These glycopeptide signals are related by the mass differences corresponding to glycosyl residues
To determine if similar enrichment efficiency could be extended to non-sialylated glycopeptides, two other standard glycoproteins, asialofetuin and recombinant EPO produced in BHK21 cells, were additionally investigated. Most tryptic glycopeptides from these two proteins were non-sialylated and a significant proportion of the glycopeptides from EPO was sulfated [36]. The MALDI-MS profiles of the tryptic peptides from asialofetuin and EPO showed that glycopeptide signals were clearly observed only after enrichment by NH2-functionalized nanoparticles (Figs. 2D and 3B) when most of the dominant signals attributed to non-glycopeptides (Figs. 2C and 3A) were no longer observed. In addition, sulfated glycopeptides from EPO could be detected in negative ion mode (Fig. 3C), corresponding to a mass shift of 78 u higher than the m/z values of their non-sulfated counterparts in positive ion mode (Fig. 3B). Collectively, the data demonstrated that the NH2-based nanoparticle approach could non-selectively enrich all major classes of glycopeptides carrying neutral, sialylated or sulfated N-glycans at femtomole level prior to direct MS applications, with high specificity against non-glycosylated peptides. For tryptic peptides from single protein or simple protein mixtures, an additional advantage is that the glycopeptide-bound nanoparticles can be spotted onto MALDI target plate and the bound glycopeptides directly desorbed for MS and MS/MS analysis. This is not only time saving but also increases the overall detection sensitivity by minimizing sample loss incurred by elution and microscale sample transfers. Furthermore, additional enzymatic treatment can be readily performed in situ on the glycopeptides bound to the nanoparticles to derive extra-structural information.

MALDI-MS spectra of EPO tryptic peptides without enrichment (A), extracted with NH2-functionalized Fe3O4 nanoparticles and detected in positive ion linear mode (B), or negative ion linear mode (C). In negative ion mode, the glycopeptide signals detected could be assigned as carrying additional sulfate modification, relative to the corresponding glycopeptide signals detected in positive ion mode. These were further confirmed by de-N-glycosylation with further MALDI-MS/MS sequencing of both the de-N-glycosylated peptides and the released N-glycans, as shown in Fig. 4. Symbols used are as previously described
Enzymatic deglycosylation for N-glycosylation sites determination
PNGase F is widely used for de-N-glycosylation to determine site-specific glycosylation occupancy by observing the characteristic 0.98 Da mass shift due to deamidation of Asparagine (N) to Aspartic acid (D) during the enzymatic process. Application to enriched glycopeptides bound to functionalized nanoparticles was therefore expected to allow subsequent direct MALDI-MS analysis of either the de-N-glycosylated peptides or the released glycans by judicious choice of matrix. As demonstrated here with the enriched glycopeptides, the de-N-glycosylated peptides from fetuin (m/z at 1,741, 3,017, and 3,672) (ESM Fig. S2) and EPO (m/z at 2,360, and 2,805) (Fig. 4A) could indeed be efficiently detected using the CHCA matrix and further selected for MALDI MS/MS to confirm the peptide sequence and glycosylation site (ESM Fig. S2 and S3). By using the DHB matrix instead, the released N-glycans could be detected directly (Fig. 4B), or after permethylation (Fig. 4C) to increase sensitivity of detection, particularly to stabilize the sialic acids for analysis in positive ion mode, or to selectively detect the sulfated glycans in negative ion mode [36]. Likewise, the detected N-glycans could be further sequenced to confirm structures by additional MALDI-MS/MS analysis (data not shown).

MALDI-MS spectra of the de-N-glycosylated peptides (A) and the released N-glycans (B, C) from the PNGase F treated, NH2-nanoparticle enriched EPO glycopeptides, acquired in reflectron mode. The de-N-glycosylated peptides were detected using CHCA as matrix (A), whereas both the native (B) and permethylated N-glycans (C) were detected by using DHB as matrix. Residues marked with D indicate the N-glycosylation sites with conversion of Asn to Asp after PNGase F treatment. Both the de-N-glycosylated peptide sequences (ESM Fig. S3), and the structures of the released N-glycans were further validated by MALDI-MS/MS analyses (data not shown)
Elution of glycopeptides from functionalized nanoparticles
Although MALDI-MS is suitable for high-throughput survey mapping of glycopeptides derived from single or few glycoproteins based on the characteristic glycosyl residue mass differences incurred by the usual glycosylation heterogeneity, more precise information on site-specific glycosylation is usually difficult to obtain. Most of the current generation of MALDI-instruments only allow efficient MS/MS analysis of the deglycosylated peptides and released glycans but not the intact, large, poorly resolved glycopeptides. The problems are more acute when one tackles the full complexity of enriched glycopeptides in shotgun glycoproteomic analysis, which would necessitate additional LC-separation most conveniently implemented via advanced LC-MS/MS analysis. For such applications, efficient elution of the captured glycopeptides from the NH2-functionalized nanoparticles in buffer compatible to LC-MS/MS analysis is a prerequisite to increase its versatility as a general glycopeptide enrichment tool.
Following the extensive washes to get rid of most of the non-glycopeptides, repeated sonication in a 50% acetonitrile/2% NH4OH solution was found to be sufficient in disrupting the hydrophilic interactions between glycopeptides and the NH2-functionalized nanoparticles. Tested against glycopeptides derived from fetuin (ESM Fig. S1d) and other glycoproteins, the MALDI-MS signal intensity and signal-to-noise ratios afforded by the eluted glycopeptides appeared to be not as good as those observed when the nanoparticle-bound glycopeptides were desorbed directly from the MALDI plate (Fig. 2B). This suggested that direct desorption either improved overall MALDI ionization and detection efficiency or minimized sample loss. However, in comparison with similar MALDI-MS analysis of non-enriched sample (ESM Fig. S1a), the glycopeptide signal cluster at around m/z 6,000 was detected at better signal-to-noise ratio albeit a small reduction in the absolute MS signal intensity registered. On the other hand, as also commonly observed in many other cases, the previously non-detected glycopeptide signal cluster at around m/z 4,300 now became clearly visible. This would imply that any loss during the elution process was more than compensated by gain in MS signal strength and quality by the enrichment process. A more practical assessment of the enrichment performance was undertaken by comparing the number of glycopeptide precursors that were successfully selected for MS/MS analysis in an LC-MS/MS run programmed for data-dependent acquisition of the three most intense precursor ions. Glycopeptides were manually identified and distinguished from non-glycosylated peptides by virtue of the characteristic glyco-oxonium ions at m/z 204.08 for HexNAc, m/z 292.10 for NeuAc, and m/z 366.14 for Hex-HexNAc. Without the enrichment step, a direct LC-MS/MS analysis of a tryptic digest of fetuin resulted in only four glycopeptide precursor ions being triggered for MS/MS. In contrast, more than twice of this number of glycopeptides were successfully selected if the sample was first subjected to enrichment (Table 1).
Enrichment of glycopeptides from mouse uterine luminal fluid
To further evaluate the practical utility and gain afforded by our glycopeptides-enrichment method in shotgun glycoproteomics, the NH2-functionalized nanoparticles were applied to a tryptic digest of mouse ULF, previously reported to carry sialylation and multiple fucosylation [37]. As expected, molecular ion signals contributed by non-glycosylated peptides dominated all survey MS scans acquired in an LC-MS/MS analysis of the non-enriched sample (Fig. 5A), whereas the glycopeptide signals became prominent (Fig. 5B) and more efficiently selected for MS/MS acquisition after enrichment. As a convenient measure of the glycopeptide enrichment efficiency for such a complex biological sample, the positively identified de-N-glycosylated peptides in a separate LC-MS/MS analysis of the enriched sample after PNGase F treatment were enumerated. This was further compared with the corresponding number derived from parallel analysis of the same sample enriched instead by the use of Sepharose CL-4B [15] resin and similarly de-N-glycosylated by PNGase F.

Comparative LC-MS survey profiling of the eluting tryptic peptides of mouse uterine luminal fluid samples before (a) and after (b) NH2-nanoparticle enrichment, as represented respectively by the corresponding single MS scans. The multifucosylated glycopeptide signal clusters were indicated by mass intervals corresponding to fucosyl residue (red triangles) difference, which were clearly more prominent and occurring as the base peaks after enrichment. The glycopeptides signals here were subsequently identified as being derived from lactotransferrin. Other than the Fuc-related molecular ions, oxonium ion signals corresponding to Fuc1HexHexNAc and Fuc2HexHexNAc epitopes were also detected at the lower m/z range. BPI, base peak intensity
It was found that without enrichment, only 37 out of 209 (18%) unique peptides were identified as de-N-glycosylated peptides carrying the characteristic Asn to Asp conversion, as confirmed by MS/MS analysis (Fig. 6A). By incorporating a glycopeptide enrichment step, the probability to detect de-N-glycosylated peptides increased more than twice, for both the NH2-based nanoparticle (43%) and the Sepharose CL-4B (40%) methods. Although both approaches enriched glycopeptides by hydrophilic interaction, a total of 18 (NH2-nanoparticles) and 8 (Sepharose CL-4B) unique peptides could only be detected by one of the two specific approaches (Fig. 6B; ESM Table S1) while 43 peptides could be identified by both methods. Translating into the number of glycoproteins identified based on one or more unique de-N-glycosylated peptides, 24 out of the 58 proteins identified (41%) without the glycopeptide enrichment step were considered glycoproteins (Fig. 6C). This percentage of glycoprotein identification increased significantly to 77% and 70% for samples enriched with NH2-nanoparticles and Sepharose CL-4B, respectively.

A comparison of the number of de-N-glycosylated peptides identified using different glycopeptide enrichment approaches. a Histograms showing the total number of identified non-glyosylated peptides (dark gray) versus de-N-glycosylated peptides containing N-glycosylation sites with Asn to Asp conversion after PNGase F treatment (light gray), for samples without glycopeptide enrichment, and those enriched by either NH2-nanoparticles or Sepharose CL-4B. b Venn diagram plot describes the number of unique de-N-glycosylated peptides identified in each approach. A detailed list for the peptide sequences and N-glycosylation sites identified are provided in ESM Table S1. c Histograms showing the total number of glycoproteins identified by unique de-N-glycosylated peptides (light gray) versus other proteins identified without any de-N-glycosylated peptides (dark gray), for each of the three samples
It is clear from the data obtained that our NH2-functionalized nanoparticles performed favorably against the other hydrophilic interaction-based glycopeptide enrichment method selected here for direct comparative evaluation and, in fact, against any other non-selective glycopeptide enrichment methods reported in the literature [10, 38, 39]. None of the existing methods achieved absolute specificity and therefore it is not surprising and commonly understood that different approaches tend to produce non-completely overlapping subsets of glycopeptides. The number of unique glycopeptides actually identified will further depend on the complexity and dynamic range of abundance exhibited by the individual glycoproteomic samples. A better enrichment performance in terms of the percentage of identified de-N-glycosylated peptides versus non-glycopeptides was actually obtained when the same experimental procedures using NH2-nanoparticles were applied to a more complex mouse serum sample (ESM Fig. S4). Approximately 5-fold increase in the percentage of identified de-N-glycosylated peptides was achieved compared with 2.4-fold increment for ULF. It should be further noted that each de-N-glycosylated peptide actually represents many glycopeptides (>10 on average) in the original sample due to natural occurrence of glycoforms, all of which collapse into a single common de-N-glycosylated peptide backbone. On the other hand, each of the identified non-glycosylated peptide represents only one peptide. Thus the actual enrichment factor, if all the peptides and glycopeptides could be detected and counted, would have been much greater. It is, however, highly sample dependent and difficult to be extrapolated from one single glycoprotein or biological sample to another.
Conclusions
Both glycan-selective and non-selective enrichment methods for glycopeptides are required in protein glycosylation and glycoproteomic analysis, depending on the overall aims. We have successfully developed an NH2-functionalized nanoparticle-based, non-selective glycopeptide enrichment method that performs equally well if not better than others. Similar to all other hydrophilic interaction-based glycopeptide capture methods, specificity is highly affected by the nature of both the glycan moiety and the attached peptide backbone, which itself varies in length and hydrophilicity depending on its amino acid sequence. We have demonstrated that the same peptides carrying neutral, sialylated and sulfated glycans could all be efficiently enriched in simple steps for MS analysis. However, as in the case for phosphopeptide enrichment, a combinatorial approach using more than one affinity capture tools will be needed if one aims for comprehensiveness [40]. More importantly, the nanoparticle-based approach allows both direct analysis by MALDI-MS, as well as LC-MS/MS after further elution. The former can be further coupled with in situ enzymatic de-N-glycosylation and analyses of both peptides and released glycans can be effected, with and without additional chemical derivatization, which is most productive for relatively simple protein glycosylation analysis. It significantly reduces the analysis time and sample loss, and allows extracting glycopeptides at femtomole level. The latter, on the other hand, is a requisite for any shotgun glycoproteomic analysis and is not limited by capacity since the number of functionalized nanoparticles used can be readily adjusted. In either case, sample manipulation is minimal, and we have developed elution conditions that do not introduce high salt content and therefore facilitate high efficiency direct LC-MS/MS analysis. The enrichment process itself is rapid, taking less than 10 min to complete if the samples are directly spotted on MALDI plate. It takes another 10 min or so to elute off the enriched glycopeptides and ready for LC-MS/MS applications. Taken together, the enrichment method based on NH2-functionalized nanoparticles will be a very useful addition to the armory of tools enabling better implementation of glycoproteomics.
Abbreviations
- Fuc:
-
Fucose
- Gal:
-
Galactose
- GlcNAc:
-
N-Acetylglucosamine
- Hex:
-
Hexose
- HexNAc:
-
N-Acetylhexosamine
- HILIC:
-
Hydrophilic interaction liquid chromatography
- MS:
-
Mass spectrometry
- Neu5Ac:
-
N-Acetylneuraminic acid
- ULF:
-
Uterine luminal fluid
References
Paulson JC, Blixt O, Collins BE (2006) Sweet spots in functional glycomics. Nat Chem Biol 2(5):238–248
Varki A (2008) Sialic acids in human health and disease. Trends Mol Med 14(8):351–360
Hakomori S (1996) Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res 56(23):5309–5318
Zaia J (2010) Mass spectrometry and glycomics. OMICS 14(4):401–418
Hart GW, Copeland RJ (2010) Glycomics hits the big time. Cell 143(5):672–676
Pan S, Chen R, Aebersold R, Brentnall TA (2011) Mass spectrometry based glycoproteomics—from a proteomics perspective. Mol Cell Proteomics 10 (1):R110 003251
Wuhrer M, Catalina MI, Deelder AM, Hokke CH (2007) Glycoproteomics based on tandem mass spectrometry of glycopeptides. J Chromatogr 849(1–2):115–128
Madera M, Mechref Y, Klouckova I, Novotny MV (2007) High-sensitivity profiling of glycoproteins from human blood serum through multiple-lectin affinity chromatography and liquid chromatography/tandem mass spectrometry. J Chromatogr 845(1):121–137
Johansen E, Schilling B, Lerch M, Niles RK, Liu H, Li B, Allen S, Hall SC, Witkowska HE, Regnier FE, Gibson BW, Fisher SJ, Drake PM (2009) A lectin HPLC method to enrich selectively-glycosylated peptides from complex biological samples. J Vis Exp (32):1398
Zielinska DF, Gnad F, Wisniewski JR, Mann M (2010) Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141(5):897–907
Larsen MR, Jensen SS, Jakobsen LA, Heegaard NH (2007) Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol Cell Proteomics 6(10):1778–1787
Alvarez-Manilla G, Atwood J 3rd, Guo Y, Warren NL, Orlando R, Pierce M (2006) Tools for glycoproteomic analysis: size exclusion chromatography facilitates identification of tryptic glycopeptides with N-linked glycosylation sites. J Proteome Res 5(3):701–708
Snovida SI, Bodnar ED, Viner R, Saba J, Perreault H (2010) A simple cellulose column procedure for selective enrichment of glycopeptides and characterization by nano LC coupled with electron-transfer and high-energy collisional-dissociation tandem mass spectrometry. Carbohydr Res 345(6):792–801
Li J, Li X, Guo Z, Yu L, Zou L, Liang X (2011) Click maltose as an alternative to reverse phase material for desalting glycopeptides. Analyst 136(19):4075–4082
Wada Y, Tajiri M, Yoshida S (2004) Hydrophilic affinity isolation and MALDI multiple-stage tandem mass spectrometry of glycopeptides for glycoproteomics. Anal Chem 76(22):6560–6565
Wuhrer M, Koeleman CA, Hokke CH, Deelder AM (2005) Protein glycosylation analyzed by normal-phase nano-liquid chromatography–mass spectrometry of glycopeptides. Anal Chem 77(3):886–894
Takegawa Y, Deguchi K, Keira T, Ito H, Nakagawa H, Nishimura S (2006) Separation of isomeric 2-aminopyridine derivatized N-glycans and N-glycopeptides of human serum immunoglobulin G by using a zwitterionic type of hydrophilic-interaction chromatography. J Chromatogr A 1113(1–2):177–181
Mysling S, Palmisano G, Hojrup P, Thaysen-Andersen M (2010) Utilizing ion-pairing hydrophilic interaction chromatography solid phase extraction for efficient glycopeptide enrichment in glycoproteomics. Anal Chem 82(13):5598–5609
Parker BL, Palmisano G, Edwards AV, White MY, Engholm-Keller K, Lee A, Scott NE, Kolarich D, Hambly BD, Packer NH, Larsen MR, Cordwell SJ (2011) Quantitative N-linked glycoproteomics of myocardial ischemia and reperfusion injury reveals early remodeling in the extracellular environment. Mol Cell Proteomics 10 (8):M110 006833
Larsen MR, Hojrup P, Roepstorff P (2005) Characterization of gel-separated glycoproteins using two-step proteolytic digestion combined with sequential microcolumns and mass spectrometry. Mol Cell Proteomics 4(2):107–119
Alley WR Jr, Mechref Y, Novotny MV (2009) Use of activated graphitized carbon chips for liquid chromatography/mass spectrometric and tandem mass spectrometric analysis of tryptic glycopeptides. Rapid Commun Mass Spectrom 23(4):495–505
Hua S, Nwosu CC, Strum JS, Seipert RR, An HJ, Zivkovic AM, German JB, Lebrilla CB (2012) Site-specific protein glycosylation analysis with glycan isomer differentiation. Anal Bioanal Chem (in press)
Zhang H, Li XJ, Martin DB, Aebersold R (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol 21(6):660–666
Gao M, Deng C, Zhang X (2011) Magnetic nanoparticles-based digestion and enrichment methods in proteomics analysis. Expert Rev Proteomics 8(3):379–390
Sparbier K, Koch S, Kessler I, Wenzel T, Kostrzewa M (2005) Selective isolation of glycoproteins and glycopeptides for MALDI-TOF MS detection supported by magnetic particles. J Biomol Tech 16(4):407–413
Sparbier K, Asperger A, Resemann A, Kessler I, Koch S, Wenzel T, Stein G, Vorwerg L, Suckau D, Kostrzewa M (2007) Analysis of glycoproteins in human serum by means of glycospecific magnetic bead separation and LC-MALDI-TOF/TOF analysis with automated glycopeptide detection. J Biomol Tech 18(4):252–258
Lee YC, Block G, Chen H, Folch-Puy E, Foronjy R, Jalili R, Jendresen CB, Kimura M, Kraft E, Lindemose S, Lu J, McLain T, Nutt L, Ramon-Garcia S, Smith J, Spivak A, Wang ML, Zanic M, Lin SH (2008) One-step isolation of plasma membrane proteins using magnetic beads with immobilized concanavalin A. Protein Expr Purif 62(2):223–229
Chu ST, Huang HL, Chen JM, Chen YH (1996) Demonstration of a glycoprotein derived from the 24p3 gene in mouse uterine luminal fluid. Biochem J 316(Pt 2):545–550
Lin SY, Chen YY, Fan YY, Lin CW, Chen ST, Wang AH, Khoo KH (2008) Precise mapping of increased sialylation pattern and the expression of acute phase proteins accompanying murine tumor progression in BALB/c mouse by integrated sera proteomics and glycomics. J Proteome Res 7(8):3293–3303
Qhobosheane M, Santra S, Zhang P, Tan W (2001) Biochemically functionalized silica nanoparticles. Analyst 126(8):1274–1278
Bruce IJ, Sen T (2005) Surface modification of magnetic nanoparticles with alkoxysilanes and their application in magnetic bioseparations. Langmuir 21(15):7029–7035
Lee CL, Hsiao HH, Lin CW, Wu SP, Huang SY, Wu CY, Wang AH, Khoo KH (2003) Strategic shotgun proteomics approach for efficient construction of an expression map of targeted protein families in hepatoma cell lines. Proteomics 3(12):2472–2486
Dell A, Reason AJ, Khoo KH, Panico M, McDowell RA, Morris HR (1994) Mass spectrometry of carbohydrate-containing biopolymers. Methods Enzymol 230:108–132
Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M (2005) Parts per million mass accuracy on an orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics 4(12):2010–2021
Lee BS, Krishnanchettiar S, Lateef SS, Lateef NS, Gupta S (2005) Characterization of oligosaccharide moieties of intact glycoproteins by microwave-assisted partial acid hydrolysis and mass spectrometry. Rapid Commun Mass Spectrom 19(18):2629–2635
Yu SY, Wu SW, Hsiao HH, Khoo KH (2009) Enabling techniques and strategic workflow for sulfoglycomics based on mass spectrometry mapping and sequencing of permethylated sulfated glycans. Glycobiology 19(10):1136–1149
Kuo CW, Chen CM, Lee YC, Chu ST, Khoo KH (2009) Glycomics and proteomics analyses of mouse uterine luminal fluid revealed a predominance of Lewis Y and X epitopes on specific protein carriers. Mol Cell Proteomics 8(2):325–342
Calvano CD, Zambonin CG, Jensen ON (2008) Assessment of lectin and HILIC based enrichment protocols for characterization of serum glycoproteins by mass spectrometry. J Proteomics 71(3):304–317
McDonald CA, Yang JY, Marathe V, Yen TY, Macher BA (2009) Combining results from lectin affinity chromatography and glycocapture approaches substantially improves the coverage of the glycoproteome. Mol Cell Proteomics 8(2):287–301
Bodenmiller B, Mueller LN, Pedrioli PG, Pflieger D, Junger MA, Eng JK, Aebersold R, Tao WA (2007) An integrated chemical, mass spectrometric and computational strategy for (quantitative) phosphoproteomics: application to Drosophila melanogaster Kc167 cells. Mol Biosyst 3(4):275–286
Acknowledgments
This work was supported by Taiwan NSC grants 94-3112-B-001-009-Y and 95-3112-B-001-014 to the NRPGM Core Facilities for Proteomics located at the Institute of Biological Chemistry, Academia Sinica. The LTQ-Orbitrap data were additionally acquired at the Academia Sinica common mass spectrometry facilities, also at the Institute of Biological Chemistry. We are grateful to Dr. Sin-Tak Chu (Institute of Biochemical Sciences, National Taiwan University) for providing the mouse uterine luminal fluid samples.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1.65 mb)
Rights and permissions
About this article
Cite this article
Kuo, CW., Wu, IL., Hsiao, HH. et al. Rapid glycopeptide enrichment and N-glycosylation site mapping strategies based on amine-functionalized magnetic nanoparticles. Anal Bioanal Chem 402, 2765–2776 (2012). https://doi.org/10.1007/s00216-012-5724-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-5724-1