
Abstract
Fast capillary electrophoresis–mass spectrometry measurements under counter-electroosmotic analyte migration conditions are presented. Efficient separations of a homologous series of six hyaluronan oligosaccharides (comprising 1–6 hyalobiuronic acid moieties) could be completed in 65 s. Separations were achieved in short-length fused silica capillaries under high electric field strengths of up to 1.25 kV·cm−1. Capillary inner diameters ranging from 5 to 50 μm were investigated, resulting in an optimal value of 15 μm. The influence of capillary dimensions and buffer composition on separation efficiency and sensitivity are discussed. Optimal separations were achieved using a 28 cm × 15 μm capillary, a separation high voltage of 35 kV, a background electrolyte of 25 mM ammonium acetate adjusted to pH 8.5, and negative ionization mode. The optimized method was successfully applied to a bovine testicular hyaluronidase digest of hyaluronan. Only minimal sample pretreatment for protein-containing samples is required. The simple manual injection procedure and fast separations allow for a sample throughput of 35 samples per hour.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Capillary electrophoresis (CE) continues to be an important separation method in biochemical sciences, owing to its ability to efficiently separate charged analytes. In recent years, however, liquid chromatography has surpassed CE in separation speed and efficiency, mainly due to the introduction of small particle columns and corresponding instrumentation. CE analyses are particularly time-consuming when the analytes of interest are negatively charged under normal (cathodic) electroosmotic flow (EOF) conditions. This counter-electroosmotic migration direction, combined with current capillary electrophoresis–mass spectrometry (CE–MS) instrumentation, typically leads to analysis times between 30 and 60 min.
Hyaluronan, a major high molecular weight component of the extracellular matrix, is composed of 1 → 4-linked β-d-glucuronic acid-(1 → 3)-β-N-acetyl-d-glucosamine disaccharide units. The biopolymer and the corresponding degradation products, oligomers resulting from enzymatic cleavage by hyaluronidases, are supposed to have various size-dependent and, in part, contrary effects on physiological and pathophysiological processes [1] such as tissue regeneration and wound healing [2, 3], inflammation [4], as well as tumor cell proliferation, invasion, and metastasis [5–7]. The kinetics of hyaluronan metabolism is widely unknown due to the lack of fast, selective, and sensitive bioanalytical methods able to monitor the enzymatic degradation of hyaluronan.
Although CE analysis of hyaluronan (high molecular weight polymer) was reported by Grimshaw et al. in the 1990s [8, 9], interest in hyaluronan oligosaccharide analysis is more recent. Analytical protocols reported recently employ UV detection and take about 40 min, with an additional 15 min of capillary conditioning [10, 11]. The resulting total analysis time of approximately 1 h is prohibitive especially for an enzymological characterization of hyaluronidases.
Conventional CE instrumentation requires capillaries of at least 75 cm length in order to be hyphenated with mass spectrometry and, due to practical reasons, the achievable high voltage is limited to about 30 kV. This results in low field strength, causing long analysis times. Both the application of high electrical field strengths [12–15] and the use of short-length capillaries [16–21] for CE experiments have been demonstrated.
Capillary material with an inner diameter (ID) below 50 μm has been applied, in order to work with very small injection volumes in conjunction with single-cell analysis. The high sensitivity of amperometric detection was used in most of these studies [22–30], whereas recent work by the group of Sweedler has been conducted with mass spectrometry (MS) detection [31].
Recently, we have shown fast and efficient CE–MS analysis in 30 s of cationic model compounds using short capillaries of 28 cm length, ID ranging from 75 to 5 μm, and electrical field strengths of 1.25 kV·cm−1 [32].
In this work, we report on an optimized CE–electrospray ionization–time-of-flight mass spectrometry (CE–ESI–TOF-MS) method for the analysis of hyaluronan oligomer mixtures. The influence of capillary length, capillary ID, and different buffer compositions are shown. The optimized method is applied to a hyaluronan digest where the molecular information obtained from mass spectrometric detection is indispensable in obtaining a complete overview over all species present in the samples.
Materials and methods
Instrumentation
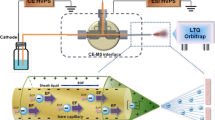
All CE experiments were performed using a laboratory-made CE instrument consisting of a high voltage power supply (model HCN 7E-35000, F.u.G. Elektronik, Rosenheim-Langenpfunzen, Germany), a control unit, and a manual sample injection system. The separation capillaries were coupled to a microTOF-MS (Bruker Daltonik, Bremen, Germany) using a coaxial sheath-liquid sprayer (Agilent Technologies, Waldbronn, Germany).
Materials and chemicals
Fused silica capillaries of 360 μm outer diameter and 50, 25, 15, 10, and 5 μm inner diameters from Polymicro Technologies (Phoenix, AZ, USA) were purchased from Optronis (Kehl, Germany). Water purified with either an Astacus system (MembraPure, Bodenheim, Germany) or a Milli-Q system (Millipore, Eschborn, Germany) and HPLC-MS-grade 2-propanol (Carl Roth, Karlsruhe, Germany) were used throughout this study. Perfluoroheptanoic acid, caesium hydrogen carbonate, and sodium hydroxide solution (50%, w/w) were from Sigma Aldrich (Munich, Germany). Ammonium acetate (NH4OAc), ammonia solution (50%, v/v), and formic acid were from Merck (Darmstadt, Germany). Background electrolyte (BGE) solutions were freshly prepared from an NH4OAc solution of given concentration, pH-adjusted with ammonia, and filtered through 0.2 μm Millex-GP syringe filter units (Millipore, Cork, Ireland).
Oligosaccharide standards (oligoHA by Hyalose, Oklahoma City, USA) were purchased from AMSBIO (Abingdon, UK) and used at a concentration of 20 μM for systematic method development. For digestion with bovine testicular hyaluronidase (BTH), Hyalo-oligo, a hyaluronan preparation with a molecular weight below 10 kDa, kindly provided by Kewpie (Tokyo, Japan), served as substrate. BTH (Neopermease®) was kindly provided by Sanabo (Vienna, Austria). Bovine serum albumin was purchased from Serva (Heidelberg, Germany). Sodium chloride for preparation of the incubation buffer was from VWR (Haasrode, Belgium); citric acid and disodium hydrogen phosphate were from Merck (Darmstadt, Germany). Phenex-NY 4 mm syringe filters 0.2 μm (Phenomenex, Aschaffenburg, Germany) were used for filtration of hyaluronan digests prior to injection.
Manual sample injection technique
The manual sample injection protocol used was the same as reported in Grundmann and Matysik [32]. Briefly, for capillary IDs of 50 and 25 μm, the capillary was manually transferred from the buffer to the sample vial for a given time interval, and then back. For capillary IDs of 15, 10, and 5 μm, additional pressure was applied to the sample vial. The laminar flow through the capillary under injection conditions was determined, and the injection period as well as the additional pressure was adjusted to yield injection plug lengths corresponding to 2% of the capillary length.
MS method development
Prior to the systematic development of the CE method, instrumental parameters with respect to the electrospray interface and the TOF-MS were optimized. The analytes showed best response in negative ion mode with an acidic sheath liquid (2-propanol/water/formic acid 50:50:0.2, v/v). Further parameters and their optimal values were as follows: sheath liquid flow rate (8 μL·min−1), nebulizer gas pressure (1.0 bar), voltages of transfer capillary (5 kV), transfer capillary exit (−100 V), skimmer 1 (−33 V), and hexapole RF (800 V). Ion transfer optics was optimized for the desired mass range of 250–1,600 m/z using sodium formate clusters. With the exception of experiments with a focus on the mass range above 1,300 m/z, where caesium perfluoroheptanoic acid clusters were used [33], mass calibration was normally done using sodium formate clusters.
For the calculation of extracted ion traces of the analytes, only m/z values which had an abundance of at least 20% of the species’ base peak were used. This was found to yield best signal-to-noise (S/N) ratios as opposed to summing up all possible isotope signals. The same threshold was applied when inclusion of species arising from adduct formation (mainly sodium adducts) was decided.
EOF marker
Since the analytes of interest were in the mass range of 250 m/z and above, and the TOF-MS was optimized to give optimal signal intensity in this region, commonly applied EOF markers of low mass could not be used. A further constraint in the choice of an EOF marker was the aqueous BGE, in which it has to be readily soluble, while being uncharged under separation conditions and ionizable in the ESI interface. Pittman et al. could show that rhodamine B shows zero net charge in the pH range of 8.3–9.0 and can be used as EOF marker [34]. However, rhodamine B is only ionized in positive ion mode, while the analytes require the use of negative ion mode. Therefore, it was necessary to switch between positive and negative ion modes while recording analyses. Since this polarity change takes approximately 20 s, it was impossible to record both EOF marker and analytes during final fast CE–MS experiments. Rhodamine B was added to analyte solutions yielding a concentration of 1 μM.
Digest of hyaluronan with BTH
The protocol for a hyaluronan digest was essentially performed according to the colorimetric method for determination of hyaluronidase activity [35, 36] and the conditions used by Hofinger et al. for CE analysis [10, 11]. McIlvaine’s buffer was prepared by mixing a 0.2 M solution of disodium hydrogen phosphate and a 0.1 M solution of citric acid, each containing 0.1 M of sodium chloride, to reach a pH value of 5.0. The incubation mixture consisted of 400 μl of buffer, 200 μl of water, 100 μl of bovine serum albumin (BSA) solution (0.2 mg/ml), 100 μl of hyaluronan solution (5 mg/ml), and 100 μl of BTH (400 IU/ml according to supplier’s information) dissolved in BSA solution (0.2 mg/ml). After the addition of BTH, the mixture was incubated for 2 and 24 h, respectively, at 37 °C. The reaction was stopped by boiling for 15 min, and denaturated protein was removed by centrifugation at 13,000 · g for 10 min. Before analysis, samples were filtered through a 0.2 μm syringe filter.
To investigate possible matrix effects, the sample was prepared as described above, except that oligosaccharide standards were used instead of hyaluronan, and BSA solution (0.2 mg/ml) was used instead of enzyme solution.
Results and discussion
Hyaluronan and hyaluronan oligomeres
Hyaluronan is a biopolymer of glycosaminoglycan structure. The repeating disaccharide unit (hyalobiuronic acid) consists of d-glucuronic acid and d-N-acetylglucosamine. The pK a of the carboxylic acid groups range between 3 and 4. [37] Figure 1 shows the monomer unit in its charge state under both physiological and the separation conditions as applied in this study, as well as the three oligosaccharides used for method development. Peak numbering in all electropherograms will refer to the number of monomers, n.

Structure of hyaluronan oligosaccharides. Peak numbering in all following figures refers to the number of disaccharide units, n
Starting from a typical set of CE method parameters for hyaluronan oligosaccharide analysis, we optimized capillary length, capillary ID, buffer concentration, and buffer pH. It should be noted that different combinations of the parameters investigated were explored, but only the relevant ones are presented in a logical order. Peak height and width (as width at half maximum) are used to compare separations.
Capillary length
Separation in CE is based on differences in electrophoretic mobility. In the context of fast CE determinations and, hence, short residence times in the separation capillary, an increased electrical field strength should be applied in order to ensure high separation efficiency.
Keeping the separation high voltage constant at 35 kV, Fig. 2 shows the effect of decreasing the capillary length and, hence, increasing the field strength. This leads to three effects: (1) The separation time decreases from over 7 to 1.3 min; (2) The separation efficiency is almost not adversely affected; (3) Signal intensities increase initially and then stay constant. The latter result is of particular interest, since the relative injection amount is kept constant (2% of the capillary length), but the absolute injection amount decreased with capillary length. A capillary length of 28 cm and a high voltage of 35 kV (corresponding to 1.25 kV·cm−1) were chosen for all further experiments.

CE–MS determinations of the three hyaluronan oligosaccharides at different electric field strength but constant separation high voltage of 35 kV. Mass traces shown are extracted ion traces. Ion selection for all traces is discussed in the experimental section. Analytes were 20 μM and signals numbered as in Fig. 1. Separation capillaries were of 50 μm ID and 75 (a), 50 (b), 43 (c), 30 (d), and 28 cm (e) in length. BGE was 25 mM NH4OAc of pH 8.5
Capillary ID
We have recently shown that the usage of capillaries with smaller ID can result in increased separation efficiency [32]. In addition, BGE with a much higher conductivity could be employed, yielding further improvements in separation efficiency. At the same time, peak heights decreased for capillaries with ID smaller than 50 μm, based on the cationic model compounds.
In the present study concerning counter-electroosmotic (anionic) species, the latter effect is much less pronounced. As can be seen in Fig. 3, peak heights remain constant from 50 to 15 μm ID, even though the injection volume decreased by a factor of 11. For 10 and 5 μm ID, peak heights decrease noticeably. Separation efficiency improves with decreasing capillary ID. The resulting narrower peaks required an increase in the data acquisition rate from 5 to 10 Hz. While this change had no influence on S/N ratios, absolute signal intensity decreased by a factor of 2. Y axis scaling in Fig. 3 was adjusted accordingly.

Influence of capillary ID on separation. Separations at 35 kV were done in capillaries of 28 cm length (corresponding to 1.25 kV·cm−1) and 50 (a), 25 (b), 15 (c), 10 (d), and 5 μm (e) ID. BGE composition, analyte concentration, and signal numbering as in Fig. 2. Data acquisition rate was 5 Hz (a–c) and 10 Hz (d, e)
A capillary ID of 15 μm was chosen for further experiments. Out of the ID investigated, it was the smallest without noticeable peak height decrease while allowing a wider choice in BGE conductivity.
BGE composition
BGE optimization started with conditions reported for CE–UV methods [10, 11], which were adjusted to be compatible with ESI–MS.
The BGE used in this study was aqueous NH4OAc, adjusted to the desired pH. The concentration of this buffer had an unexpected effect on separations as is shown in Fig. 4. An increase in buffer concentration led to a decrease in signal intensity. Since peak areas remain constant between separations with different BGE concentrations, ion suppression can be ruled out as explanation. Peak broadening due to Joule heating is more likely; for the highest concentration, 100 mM, this can be clearly seen. For further experiments, a BGE concentration of 25 mM was chosen, since it resulted in the same peak height as a 10 mM BGE, while providing a higher buffer capacity.

Influence of BGE concentration on separation. BGE was 10 (a), 25 (b), 50 (c), and 100 mM (d) NH4OAc, adjusted with ammonia to pH 8.5. Separation capillary was of 28 cm length and 15 μm ID. Separations were done at 35 kV (corresponding to 1.25 kV · cm−1). Analyte concentration and signal numbering as in Fig. 2
The influence of BGE pH is shown in Fig. 5. An increasing pH led to sharpened signals and shorter migration times but also decreased separation efficiency. As a compromise between these parameters, a BGE pH of 8.5 was selected for further experiments.

Influence of BGE pH on separation. BGE was 25 mM NH4OAc, adjusted with ammonia to pH 7.0 (a), 7.5 (b), 8.0 (c), 8.5 (d), and 9.0 (e). Other conditions as in Fig. 4
Optimized method parameters
Capillaries of 28 cm length and 15 μm ID were found to give best results. The BGE consisted of 25 mM aqueous NH4OAc adjusted with ammonia to pH 8.5. The limits of detection for the analytes investigated were in the range of 4–7 · 10−7 M. Further figures of merit for the optimized method are summarized in Table 1. Determinations could be completed within about 70 s after injection. When including the injection protocol and manual handling, determinations can be run at 100 s intervals under optimal conditions.
No adverse effect of injecting BSA-containing samples was observed, even after repeated runs without capillary conditioning in between. Most matrix components showed very low or no electrophoretic mobility, so that the analytes were separated by about 30 s from the matrix components (see Electronic Supplementary Material Fig. S1). No influence on quantification could be observed when comparing samples containing only the analytes of interest and samples which contained BSA in addition.
Application to hyaluronan digest analysis
A sample of hyaluronan was subjected to hydrolysis by BTH as detailed in the experimental section. Direct injection of digest samples led to strong peak broadening. Dilution of 1:10 with water was found to yield best signal intensity and separation efficiency, while higher dilutions resulted in a loss of sensitivity and lower dilutions still showed peak broadening. Figure 6 shows separations of 2 and 24 h digests. In addition to the three known analytes (2, 3, and 4), a further three hyaluronan oligosaccharides could be identified, which were not previously included in the method development. The shorter digest shows two larger oligosaccharides (5 and 6), while the longer digest shows formation of the monomer (1). Figure S2 (see Electronic Supplementary Material) shows experimental and calculated mass spectra for compound 5 used for identification. The sums of peak heights within each digest show good agreement between the two samples.

CE–MS of hyaluronan digests after 2 h (a) and 24 h (b) digestion periods. Samples were diluted 1:10 prior to analysis. BGE was 25 mM NH4OAc adjusted with ammonia to pH 8.5. Separations were done at 35 kV in a 28 cm × 15 μm capillary (corresponding to 1.25 kV · cm−1). Signals 1, 5, and 6 were identified from their exact m/z values
Conclusions
In this report, we have shown that fast and efficient CE–MS analyses under counter-electroosmotic conditions can be completed in about 1 min when using short-length fused silica capillaries in conjunction with high electrical field strengths. CE–MS runs were completed in 65 s, and a sample throughput of about 35 samples per hour can be achieved, which includes sample change and a manual sample injection protocol. The injection protocol could be easily automated to allow for unattended operation.
High separation efficiency in combination with fast migration times could be achieved mainly through the use of high electrical field strengths and capillaries of short length and small ID, as well as an optimized buffer composition. A capillary length of 28 cm is the shortest that can be applied to the CE–ESI–MS setup and allowed the application of a field strength of 1.25 kV·cm−1 at a separation high voltage of 35 kV. Surprisingly, a capillary ID of 15 μm showed the best analytical performance (both separation efficiency and signal intensity) in the context of this study, while a previous study of cationic analytes in formic acid showed a significant loss of signal intensity for capillary ID smaller than 50 μm [32]. The optimal buffer composition was found to be 25 mM NH4OAc adjusted with ammonia to pH 8.5.
The method was successfully applied to hyaluronan digest samples, and additional experiments showed no deteriorating effects when injecting protein-containing samples. Therefore, only minimal sample pretreatment (filtration and dilution) is necessary. This fact combined with the short total analysis time opens the possibility for enzyme kinetic studies without the need for quenching.
Abbreviations
- BGE:
-
Background electrolyte
- BSA:
-
Bovine serum albumin
- BTH:
-
Bovine testicular hyaluronidase
- CE:
-
Capillary electrophoresis
- EOF:
-
Electroosmotic flow
- ESI:
-
Electrospray ionization
- ID:
-
Inner diameter
- MS:
-
Mass spectrometry
- NH4OAc:
-
Ammonium acetate
- TOF-MS:
-
Time-of-flight mass spectrometry
References
Asari A (2005) Glycoforum. (http://www.glycoforum.gr.jp/science/hyaluronan/HA12a/HA12aE.html)
Chen WY, Abatangelo G (1999) Wound Repair Regen 7:79–89
Noble PW (2002) Matrix Biol 21:25–29
Day AJ, De la Motte CA (2005) Trends Immunol 26:637–643
Boregowda RK, Appaiah HN, Siddaiah M, Kumarswamy SB, Sunila S, Thimmaiah KN, Mortha K, Toole B, Banerjee S (2006) J Carcinog 5:2–10
Stern R (2005) Pathol Biol 53:372–382
Stern R (2008) Semin Cancer Biol 18:275–280
Grimshaw J, Kane A, Trocha-Grimshaw J, Douglas A, Chakravarthy U, Archer D (1994) Electrophoresis 15:936–940
Grimshaw J, Trocha-Grimshaw J, Fisher W, Rice A, Smith S, Spedding P, Duffy J, Mollan R (1996) Electrophoresis 17:396–400
Hofinger ESA, Bernhardt G, Buschauer A (2007) Glycobiology 17:963–971
Hofinger ESA, Hoechstetter J, Oettl M, Bernhardt G, Buschauer A (2008) Glycoconj J 25:101–109
Hjerten S, Valtcheva L, Elenbring K, Liao JL (1995) Electrophoresis 16:584–594
Hutterer KM, Jorgenson JW (1999) Anal Chem 71:1293–1297
Palonen S, Jussila M, Porras SP, Hyötyläinen T, Riekkola ML (2001) J Chromatogr A 916:89–99
Palonen S, Jussila M, Porras SP, Hyötyläinen T, Riekkola ML (2002) Electrophoresis 23:393–399
Zhong M, Lunte S (1996) Anal Chem 68:2488–2493
Müller O, Minarik M, Foret F (1998) Electrophoresis 19:1436–1444
Matysik FM (2006) Electrochem Commun 8:1011–1015
Matysik FM, Neusüß C, Pelzing M (2008) Analyst 133:1764–1766
Opekar F, Coufal P, Štulík K (2009) Chem Rev 109:4487–4499
Kennedy RT (1999) Anal Chim Acta 400:163–180
Wallingford RA, Ewing AG (1988) Anal Chem 60:1975–1977
Olefirowicz TM, Ewing AG (1990) Anal Chem 62:1872–1876
Chien JB, Wallingford RA, Ewing AG (1990) J Neurochem 54:633–638
Olefirowicz TM, Ewing AG (1990) J Neurosci Meth 34:11–15
Kristensen HK, Lau YY, Ewing AG (1994) J Neurosci Meth 51:183–188
Kennedy RT, Oates MD, Cooper BR, Nickerson B, Jorgenson JW (1989) Science 246:57–63
Hogan BL, Yeung ES (1992) Anal Chem 64:2841–2845
Blasco S, Kortz L, Matysik FM (2009) Electrophoresis 30:1–6
Lin Y, Trouillon R, Safina G, Ewing AG (2011) Anal Chem 83:4369–4392
Lapainis T, Rubakhin SS, Sweedler JV (2009) Anal Chem 81:5858–5864
Grundmann M, Matysik FM (2011) Anal Bioanal Chem 400:269–278
König S, Fales HM (1999) J Am Soc Mass Spectrom 10:273–276
Pittman JL, Schrum KF, Gilman SD (2001) Analyst 126:1240–1247
Reissig JL, Strominger JL, Leloir LF (1955) J Biol Chem 217:959–966
Muckenschnabel I, Bernhardt G, Spruss T, Dietl B, Buschauer A (1998) Cancer Lett 131:13–20
Hascall VC, Laurent TC (1997) Glycoforum. http://www.glycoforum.gr.jp/science/hyaluronan/HA01/HA01E.html
Acknowledgments
Financial support by the Deutsche Forschungsgemeinschaft (MA 1401/7-1) and the Bayerische Forschungsstiftung (AZ-829-08) is gratefully acknowledged.
This research was supported by the Research Executive Agency (REA) of the European Union under Grant Agreement number PITN-GA-2010-264772 (ITN CHEBANA).
MR expresses his gratitude to the Studienstiftung des Deutschen Volkes for a PhD grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the ANAKON special issue with guest editors P. Dittrich, D. Günther, G. Hopfgartner, and R. Zenobi
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 170 kb)
Rights and permissions
About this article
Cite this article
Grundmann, M., Rothenhöfer, M., Bernhardt, G. et al. Fast counter-electroosmotic capillary electrophoresis–time-of-flight mass spectrometry of hyaluronan oligosaccharides. Anal Bioanal Chem 402, 2617–2623 (2012). https://doi.org/10.1007/s00216-011-5254-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-5254-2