
Abstract
Laser-induced breakdown spectroscopy (LIBS) has been employed for the analysis of slurry samples. Quantitative analysis of slurry samples is crucial and challenging. The problems associated with slurry samples include splashing, surface turbulence, and the difficulties of obtaining reproducible samples due to sedimentation. The LIBS analysis has achieved limited success due to inherent disadvantages when applied to slurry samples. In order to achieve improved measurement precision and accuracy, a spin-on-glass sampling method was evaluated. Five elements (Al, Ca, Fe, Ni, and Si) were examined in five slurry simulants containing varying amounts of each ion. Three calibration models were developed by using univariate calibration, multiple linear regression, and partial least square regression. LIBS analysis results obtained from the partial least square regression model were determined to be the best fit to results obtained from inductively coupled plasma optical emission spectroscopy analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Laser-induced breakdown spectroscopy (LIBS) is a spectroscopic technique that is widely used for qualitative and quantitative analysis of the elemental composition of solids, liquids, and gases [1–3]. One of the main advantages of using LIBS is the possibility of rapid analyte measurement with little or no sample preparation. Although LIBS may be applied to any sample forms, the use of LIBS for the direct analysis of liquid samples is still very challenging due to problems associated with splashing, aerosols above the surface, and bubbling inside the liquid. The selection of sampling configuration can greatly affect the stability and reproducibility of the LIBS measurement for liquid samples. Generally, liquid samples are used in the form of a static liquid pool, droplets or aerosols, bulk flowing liquids, or liquid streams in LIBS analysis [4–9]. Previous studies have shown that the presence of solids and their constituent materials and dissolved gases can influence the laser focus and decrease the intensity of the LIBS signal. Therefore, LIBS has achieved limited success in the quantitative analysis of liquid samples. To reduce splashing and provide better limits of detection (LODs), Schmidt et al. have performed elemental analysis of solutions via LIBS by concentrating and immobilizing metal ions into an ion exchange polymer membrane [10]; Yaroshchyk et al. have employed paper substrates for the quantitative elemental analysis of used lubricating oils [11].
Applications of LIBS to slurry samples not only involve the challenge associated with liquid sample analysis but also the additional challenge of obtaining a valid representative sample of the slurry. Michaud et al. have applied LIBS to the analysis of iron ore slurries [12–14]. To overcome the problems associated with slurry samples, they used sampler geometry with a slurry recirculator to produce a column of free-falling slurry for LIBS measurement [13]. They studied the influence of particle size and mineral phase on the analysis of iron ore slurries with LIBS and discovered the LIBS intensity was dependent upon the particle size and on the magnetite content [12]. An empirical correction equation for the calibration model was needed to correct the intensity of either iron or silicon lines due to the variation of the average particle size within the ore sample. Previously, application of LIBS as a remote, real-time analytical technique to analyze liquid radioactive wastes during the vitrification process was explored using direct sampling and slurry circulation systems [15, 16]. The problems encountered with direct analysis of wet slurry samples were sedimentation, splashing, and surface turbulence. Also, water could quench the laser plasma and suppress the LIBS signal, resulting in poor sensitivity. Investigations on the effect of water from slurries on LIBS spectra indicated that as the water content decreased, the LIBS signal was enhanced with the decrease of standard deviation. A preliminary study of the slurry samples coated on substrates for elemental analysis with LIBS has shown good correlation between spectral intensity and elemental concentration [17].
For this study, a thin-coating procedure called “spin-on-glass” was applied for LIBS analysis of slurry samples. In this approach, the moisture in the slurry sample is near completely removed before analysis, and LIBS measurements transform from slurry samples analysis to almost solid samples analysis. Several experimental configurations and detection conditions were studied to obtain optimum experimental conditions for slurry measurements. To evaluate the quantitative measurement and the reproducibility capabilities of the LIBS technique, the LIBS data of an unknown slurry sample was analyzed and compared with the results of plasma optical emission (inductively coupled plasma optical emission spectroscopy (ICPOES)) analysis.
Experimental
Apparatus
The two experimental setups used to record the LIBS signal from slurry are shown in Fig. 1. A frequency doubled, Q-switched Nd:YAG laser (Continuum Surelite III, 10 Hz, 5-ns pulse width, 9 mm diameter, 425 mJ maximum) was used as an excitation source. The laser light at 532 nm was focused on the sample surface by using a 30-cm focal length of fused silica convex lens. The size of the focused spot is about 50 μm. The energies of the laser pulse used in this work were between 8 and 44 mJ, corresponding to peak intensities of approximately 8.2 × 1010 and 4.5 × 1011 W/cm2, respectively. The emitted light from the laser-induced plasma was collected either in a backward direction or with a pick-up lens (Ocean Optics Inc. (OOI) Part No. 74-UV) aligned at a 45° with respect to the laser beam to a 100-μm optical fiber. The distance of the pick-up lens was 6 cm from the focal point of the laser beam. The collected signal was sent to a broadband spectrograph (Andor Mechelle 5000), which was integrated with the intensified charge coupled detector. The detector head has a built-in digital delay generator (DDG) that controls the gate delay and gate width. To synchronize the data acquisition with the laser pulse, the DDG is activated by a trigger pulse from the output of a DDG (DG535, Stanford Research Systems) which is triggered with the laser Q-switch output. The data acquisition and analysis were performed by using a personal computer and Andor iStar software. To obtain better signal-to-noise data, each spectrum recorded was an average of five shots. Ten spectra were collected from each sample. Measurement was performed on a rotating platform to ensure data collection from various parts of the prepared sample. LIBS data were collected for various rotation speeds from 1 to 3 rotation/min.

Schematic diagram of two experimental setups of LIBS
Sample preparation
Following literature procedures, to evaluate LIBS calibration for slurry analysis, five sludge simulants were prepared and labeled as 8F, 10F, 12F, 14F, and unknown [18]. Five analytes, aluminum, calcium, nickel, iron, and silicon, were selected for weight percent adjustments based upon original percentages found in 8F simulant composition. A sample from each simulant was submitted for ICPOES analysis. Samples from 8F, 10F, 12F, and 14F simulants were utilized for instrument calibrations for each analyte. Table 1 contains the weight percent (dry) of these components obtained from the identified sludge simulants. The unknown simulant sample contained each of the selected five components differing only in the weight percentages, but remained within the calibration ranges used in the LIBS experiments. LIBS data of the four slurry samples (i.e., 8F, 10F, 12F, and 14F) were collected and used to build calibration models. The data of the unknown sample were analyzed by using these calibration models.
LIBS experiments with liquid samples create splashing and shockwaves, which cause poor signal reproducibility. The slurry samples used in this study contained mostly water (∼80%), ferric oxide, alumina, sodium oxide, and small quantities of oxides of carbon, silicon, chromium, manganese, magnesium, etc. [18]. To solve problems related with liquid samples, we have evaluated the method known as “spin-on-glass” with double-sided tape in order to reduce water content. The slurry sample is first deposited on a substrate (glass slide with double-sided tape), then placed in a spin-coater machine (Chemat KW-4A) for spinning. Samples were coated using 380 rpm for 18 s and 2,060 rpm for 60 s. By adjusting the rotation speeds, coating thickness, sample distribution on the substrate, and moisture removal will be well controlled. When the substrate is at the lowest rotation speed, the coating thickness will be adjusted, at a higher rotation speed, the distribution of sample on the substrate and moisture removal are controlled. For the average of samples, upon completion of the spin coating, excess slurry was removed, leaving the total slurry present on the substrate about 7.8 mg. Finally, the slurry-coated substrate sample was dried on a hotplate (55 °C) for 90 s to remove the moisture from the sample completely. The remaining weight of the dried slurry sample on the substrate after moisture removal was about 2.5 mg.
Two advantages of sample preparation using this method are (1) to ensure uniform thickness (2.5–3.5 μm) of the slurry sample on the glass substrate and (2) moisture content or wetness of the sample is reduced, so low laser energy is sufficient to obtain a good signal.
Data analysis
Forty spectra from each calibration sample were used to develop the calibration model, and ten additional spectra of each calibration sample were used for validation of the calibration model. Three calibration models evaluated in this work were univariate calibration (UC), multiple linear regression (MLR), and partial least square regression (PLS-1) [19, 20]. The UC is based on a plot of the signal of an analyte spectral line as a function of analyte concentration. The calibration curves were obtained from the LIBS data of a series of known concentration samples and then used to determine the concentration of an unknown sample. To account for various factors that could affect the LIBS signal, such as interference effects caused by other components of the sample matrix or random experimental errors, the MLR was also evaluated for LIBS calibration. The peak area of each selected analyte line was extracted from the spectral data of each slurry sample and normalized to the total emission intensity. The calibration curve was obtained by fitting the linear equation below to a set of experimental data consisting of the measured peak area and known concentrations of analyte. The MLR procedures estimate a linear equation of the form:
where C x is the concentration of the element x, and A i is the peak areas of the selected analyte lines. The multiple lines can be used in the regression to improve the measurement accuracy by correcting for the matrix effect. PLS-1, a multivariate analysis using whole spectrum or selected spectral regions to develop a calibration model, was also tested in this work. Some preprocessing methods, including multiple scattering correction, normalization, and baseline correction, could be applied to the calibration data sets. This analysis method relies on simultaneously fitting the intensity of the selected spectral region and species concentration to minimize the variance of both the spectral intensity and species concentration. The GRAMS/AI PLSplus/IQ (Thermo Scientific) software was used to perform multivariate analysis for slurry data.
The predictive quality of the calibration models were evaluated by calculating the correlation coefficient R 2, root mean square error prediction (RMSEP), and relative error of prediction (REP) in the validation step with independent samples. The RMSEP values are an estimate of the absolute error of prediction for each component.
The REP is calculated as:
where x i is the true concentration of the analyte in the sample i, \( {\widehat{x}_i} \) represents the estimated concentration of the analyte in the sample i, \( {\overline x_i} \) is the mean of true concentrations in the prediction set, and n is the total number of samples used in the prediction sets.
Results and discussion
Effects of laser energy and gate delay
The backward detection (Fig. 1a) and 45° detection (Fig. 1b) configurations were used in the study of experimental parameters and LIBS calibration. The backward detection allows remote measurement but demands a complex optical geometry. In comparison with backward detection, 45° detection configuration needs simple alignment and gives high sensitivity, but it cannot be employed for remote detection applications. In a 45° detection configuration, a pick-up lens aligned 45° with the laser beam was used to directly couple the LIBS signal into the optical fiber. The LIBS spectra of spin-coated samples with both detection configurations were collected, and it was determined the sensitivity was improved (∼5–10 times) with the 45° detection configuration by using low laser energy.
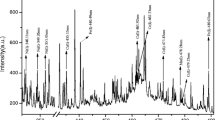
The influence of laser energies and gate delays was studied to determine the optimum experimental parameters for quantitative measurement. The relation between the intensity of analyte lines and laser energy at a constant gate delay of 0.5 μs is shown in Fig. 2a. Initially, the LIBS signal gradually increases with the increase of laser energy and becomes constant at higher laser energy. The relation between the intensity of analyte lines and gate delay at constant laser pulse energy of 22 mJ is shown in Fig. 2b. The LIBS signal decreases dramatically with the increase of gate delay time as the gate delay less than 5 μs and decreases slowly with the increase of gate delay time at higher delay times. To achieve good reproducibility, the laser-induced plasma in the measurement time frame needs to be as stable as possible. Plasma temperatures were evaluated for different experimental conditions and are shown in Fig. 3. Plasma temperatures were estimated using a Boltzmann plot of intensities of 14 spectral lines of iron between 400 and 500 nm. As the pulse energy began to rise above 22 mJ, the plasma temperature was less influenced by variations of laser pulse energy. This study indicates that laser pulse energy above 22 mJ was required to obtain reproducible measurement in LIBS analysis.

a The relation between intensity of LIBS signal and laser energy at a constant gate delay (0.5 μs). b The relation between intensity of LIBS signal and gate delay at constant laser energy (22 mJ)

Plasma temperature at different gate delays and laser pulse energies
To further improve the reproducibility of LIBS measurement and overcome the effect of pulse-to-pulse laser fluctuation, the recorded spectra were normalized by integral plasma emission [15]. This method significantly reduced the matrix influence on quantitative analyses. The measurement accuracy was also improved due to the reduction of data scattering on the calibration graphs. To test the selected analyte lines for measurement reproducibility, the five slurry samples were prepared on every day prior to running the experiment. The LIBS data of these five slurry samples were collected and normalized with the integral plasma emission. Figure 4 shows the normalized data of Ni 313.4 nm of the five slurry samples taken at a period of 4 days using experimental parameter #1 (see Table 2). As a result of this approach, the day-by-day reproducibility improved. The relative standard deviation from the measurements of 4 days was less than 4% (n = 4, each measurement was made on a different day).

Reproducibility of the normalized intensity of Ni 313.4 nm for an extended period of time
Calibration
Model validation
Three calibration models were developed and tested. Linear regression calibration assumes a linear relationship between the peak intensity (area) of the analyte line and the elemental concentration. This calibration is easy to develop; however, the relationship between signal and concentration is not always linear due to spectral interference, matrix effect, and variation of laser-induced plasma. A normalization technique was applied to the calibration data to reduce those effects. In this work, more than one line for each element was used to check the linear behavior of the calibration curves. The observed Si signals from the calibration samples have very poor signal-to-noise ratio; therefore, they were not used for this calibration. The calibration results for test data are shown in Table 3. The high R 2 values indicate that the predicted concentrations of Ni and Ca will be more reliable as compared to Fe and Al. The model has given relatively high error margin due to the effect of self-absorption for predicted concentrations of Fe and Al.
To account for various factors that could affect the LIBS signal, such as interference effects caused by other components of the sample matrix or random experimental errors, the MLR was also evaluated for LIBS calibration [21]. The peak area of each selected analyte line was extracted from the spectral data of each slurry sample and normalized to the total emission intensity using the function provided by Andor iStar software. The selected lines for the linear regression are given in Table 3. The MLR can be a good approach to predict the unknown concentration if the elemental concentration can be well correlated with the intensity of a few spectral lines. However, this method could be inefficient or inappropriate if many spectral lines are needed or if there is significant collinear relation between the spectral lines. The spectral interference, such as self-absorption, can affect the relation between spectral intensity and elemental concentration. Therefore, the selection of analyte lines in the MLR is also important. The RMSEP and REP values for validation data using the MLR model is given in Table 4 for comparison.
A multivariate calibration can use either whole spectrum or selected spectral regions to develop a calibration model. There are some preprocessing methods, such as multiple scattering correction, normalization, and baseline correction, which could be applied to the calibration data sets. PLS-1 was used in this work and relies on simultaneously fitting the intensity of the selected spectral region and the species concentration to minimize the variance of both the spectral intensity and species concentration. Because of the high Fe concentration in the calibration sample (>20 wt.%), self-absorption was found in many Fe lines. To minimize the effect of self-absorption, only selected spectral regions were used in the PLS regressions. Multiplicative scatter correction (MSC) was applied to the training set to compensate for additive and/or multiplicative effects in the spectra. The optimum number of factors in PLS to model the system without over fitting was determined to be six from a series of tests. Table 5 shows the RMSEP and REP values for validation data obtained using a PLS-1 model with a desired number of factors in the model. This model gave a reasonably good prediction for the validation set. The REP values for Al, Ca, Fe, and Ni from the model were all below 5%. The REP values for Si were 8.6%. Therefore, the prediction error for Si was expected to be higher as compared to other elements. A comparison of the test results from different calibration methods shows that the prediction error for Al, Fe, and Ni using UC, PLS-1, and MLR are comparable [22]. However, the PLS-1 model gives much less prediction error for Ca.
Comparison of LIBS and ICPOES
To evaluate the quantitative ability of LIBS, the calibration models mentioned in the previous section were tested with the data of the unknown sample taken under the same experimental conditions as a calibration data. First, the effects of experimental parameters to the LIBS analysis were evaluated with the multivariate calibration. The test experimental parameters are given in Table 2. The LIBS data of the four calibration samples were taken with these four experimental conditions and used to build calibration models. The analysis results of unknown sample from three different calibration models are shown in Table 6.
The results of Al, Fe, Ca, and Ni between different experiments were tested with F statistics and indicate that no statistically significant difference exists. The results of Si between the different experiments show larger variation as compared to other elements. To investigate this variation, the Si spectral line at 288.15 nm taken from sample 14F at different gate delay time and laser energies was compared. The Si spectral line 288.15 nm at a different gate delay time clearly shows a spectral interference from an unidentified line near 288.09 nm. The intensity ratio of Si 288.15 nm line and the interference line 288.09 nm varies with the gate delay time. It shows that the Si detection sensitivity can be improved with longer gate delay time. Because the spectral interference depends on the gate delay time and laser energy, it can explain the larger difference found in Si measurement between the different experiments.
LIBS analyses of the unknown sample were performed with UC, MLR, and PLS-1 regression and compared with ICPOES analysis. The results from PLS from different days and experimental conditions were found to be more consistent as compared to the results obtained from UC and MLR. The comparison between PLS analysis of the unknown sample and ICPOES analysis is shown in Table 7. The results for Ni were well agreed with ICPOES analysis (percent difference less than 1%). The percent differences between ICPOES and LIBS analyses for Al, Fe, and Ca are all less than 8%. The LIBS-predicted Fe concentration was found consistently lower than ICPOES analysis because the calibration samples all contain high Fe concentration (>20 wt.%), and most Fe emission lines suffered from self-absorption. The small sensitivity was produced by a less intense line for the Fe calibration curve. For the determination of high concentration elements, the measurement conducted under reduced pressure atmospheres can significantly reduce the effect of self-absorption and improve measurement quality.
The inconsistent result between LIBS and ICPOES for Si was further investigated by examining the data of all samples. One possible reason is the samples prepared for the LIBS measurement are not representative samples due to the mixing process (i.e., sample might not be thoroughly mixed). However, the repeated measurements with newly prepared samples all show the same trend. Another possible reason is the sample viscosity has affected the sample deposition on the slide during the spin-coating method. The low Si intensity from sample 12F still needs further investigation.
Conclusions
The application of LIBS to slurry samples has achieved limited success and has some inherent disadvantages. The quantitative analysis of slurry samples is crucial and challenging. A good sampling method is needed to significantly improve sensitivity and repeatability. It should also represent true sampling of the slurry. A new sampling method “spin-on-glass” was evaluated in this work. It converts liquid measurement to almost solid-sample measurement to avoid the problems associated with liquid measurement. The results of this work show this method improves the reproducibility and sensitivity of LIBS for slurry sample analysis. The inconsistent results of Si between the LIBS and ICPOES analyses indicate further study is necessary to investigate the effect of particulate size on this sampling method.
References
Cremers DA, Radziemski LJ (2006) Handbook of laser-induced breakdown spectroscopy. Wiley, West Sussex
Miziolek AW, Palleschi V, Schechter I (eds) (2006) Laser induced breakdown spectroscopy (libs): fundamentals and applications. Cambridge University Press, Cambridge
Singh JP, Thakur SN (eds) (2007) Laser induced breakdown spectroscopy. Elesvier Science B.V., Amsterdam
Rai VN, Rai AK, Yueh FY, Singh JP (2003) Optical emission from laser induced breakdown plasma of solid and liquid samples in the presence of magnetic field. Appl Opt 42:2085–2093
Samek O, Beddows DCS, Kaiser J, Kukhlevsky SV, Liska M, Telle HH, Young J (2000) Application of laser induced breakdown spectroscopy to in situ analysis of liquid samples. Opt Eng 39:2248–2262
Lui SL, Godwal Y, Taschuk MT, Tsui YY, Fedosejevs R (2008) Detection of lead in water using laser-induced breakdown spectroscopy and laser-induced fluorescence. Anal Chem 80:1995–2000
Chen Z, Li H, Liu M, Li R (2008) Fast and sensitive trace metal analysis in aqueous solutions by laser-induced breakdown spectroscopy using wood slice substrates. Spectrochim Acta B 63:64–68
Rai NK, Rai AK (2008) LIBS—an efficient approach for the determination of Cr in industrial wastewater. J Hazard Mater 150:835–838
Lazic V, Jovicevic S, Fantoni R, Colao F (2007) Efficient plasma and bubble generation underwater by an optimized laser excitation and its application for liquid analyses by laser-induced breakdown spectroscopy. Spectrochim Acta B 62:1433–1442
Schmidt NE, Goode SR (2002) Analysis of aqueous solutions by laser-induced breakdown spectroscopy of ion exchange membranes. Appl Spectrosc 56:370–374
Yaroshchyk P, Morrison RJS, Body D, Chadwick BL (2005) Quantitative determination of wear metals in engine oils using LIBS: the use of paper substrates and a comparison between single- and double-pulse LIBS. Spectrochim Acta B 60:1482–1485
Michaud D, Leclerc R, Proulx É (2007) Influence of particle size and mineral phase in the analysis of iron ore slurries by laser-induced breakdown spectroscopy. Spectrochim Acta B 62:1575–1581
Michaud D, Proulx É, Chartrand JG, Barrette L (2003) Shooting slurries with laser-induced breakdown spectroscopy: sampling is the name of the game. Appl Opt 42:6179–6183
Barrette L, Turmel S (2001) On-line iron-ore slurry monitoring for real-time process control of pellet making processes using laser-induced breakdown spectroscopy: graphitic vs. total carbon detection. Spectrochim Acta B 56:715–723
Oh SY, Yueh FY, Singh JP, Herman CC, Zeigler K (2009) Preliminary evaluation of laser induced breakdown spectroscopy for slurry samples. Spectrochim Acta B 64:113–118
Oh SY, Miller T, Yueh FY, Singh JP (2007) Comparative study of laser-induced breakdown spectroscopy measurement using two slurry circulation systems. Appl Opt 46:4020–4025
Eseller KE, Tripathi MM, Yueh FY, Singh JP (2010) Elemental analysis of slurry samples with laser induced breakdown spectroscopy. Appl Opt 49:C21–C26
Poirier MR (2005) Recipe for simulated tank 8F sludge containing no RCRA metals or halides, WSRC-TR-2005-00045 Rev.0. Westinghouse Savannah River Company, Aiken
Weisberg S (2005) Applied linear regression, 3rd edn. Wiley, Hoboken
Esbensen KH (2004) Multivariate data analysis in practice, 5th edn. Camo Inc., Oslo
Ferreira EC, Anzano JM, Milori DMBP, Ferreira EJ, Lasheras RJ, Bonilla B, Montull-Ibor B, Casas J, Neto LM (2009) Multiple response optimization of laser-induced breakdown spectroscopy parameters for multi-element analysis of soil samples. Appl Spectrosc 63:1081–1088
Braga JWB, Trevizan LC, Nunes LC, Rufini IA, Santos D, Krug FJ (2010) Comparison of univariate and multivariate calibration for the determination of micronutrients in pellets of plant materials by laser induced breakdown spectrometry. Spectrochim Acta B 65:66–74
Acknowledgements
This work is supported by US Department of Energy Cooperative Agreement no. DE-FC26-98FT 40395. The authors would like to thank Mr. Markandey M. Tripathi, Mr. Tracy Miller, and Dr. Kemal E. Eseller for help in the laboratory and Dr. Jeff Lindner for useful discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue Laser-Induced Breakdown Spectroscopy with Guest Editors Jagdish P. Singh, Jose Almirall, Mohamad Sabsabi, and Andrzej Miziolek.
Rights and permissions
About this article
Cite this article
Ayyalasomayajula, K.K., Dikshit, V., Yueh, F.Y. et al. Quantitative analysis of slurry sample by laser-induced breakdown spectroscopy. Anal Bioanal Chem 400, 3315–3322 (2011). https://doi.org/10.1007/s00216-011-4852-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-4852-3