
Abstract
An LC-MS/MS method for the determination of the atypic neuroleptic clozapine and its two main metabolites norclozapine and clozapine-N-oxide has been developed and validated for serum and urine. After addition of d4-clozapine as deuterated internal standard a fast single-step liquid–liquid extraction under alkaline conditions and with ethyl acetate as organic solvent followed. The analytes were chromatographically separated on a Synergi Polar RP column using gradient elution with 1 mM ammonium formate and methanol. Data acquisition was performed on a QTrap 2000 tandem mass spectrometer in multiple reaction monitoring mode with positive electrospray ionization. Two transitions were monitored for each analyte in order to fulfill the established identification criteria. The validation included the determination of the limits of quantification (1.0 ng/mL for all analytes in serum and 2.0 ng/mL for all analytes in urine), assessment of matrix effects (77% to 92% in serum, 21 to 78% in urine) and the determination of extraction efficiencies (52% to 85% for serum, 59% to 88% for urine) and accuracy data. Imprecision was <10%, only the quantification of norclozapine in urine yielded higher relative standard deviations (11.2% and 15.7%). Bias values were below ±10%. Dilution of samples had no impact on the correctness for clozapine and norclozapine in both matrices and for clozapine-N-oxide in serum. For quantification of clozapine-N-oxide in urine a calibration with diluted calibrators has to be used. Calibration curves were measured from the LOQ up to 2,000 ng/mL and proved to be linear over the whole range with regression coefficients higher than 0.98. The method was finally applied to several clinical serum and urine samples and a cerebro-spinal fluid sample of an intoxicated 13-month-old girl.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clozapine, a dibenzodiazepine derivative, was introduced as the first atypical antipsychotic in the 1970s. It is still regarded as one of the most effective medications for the treatment of both positive and negative signs of schizophrenia while showing practically no extrapyramidal symptoms [1]. However, a very severe, potentially life-threatening pharmacological adverse drug reaction, agranulocytosis, which occurs in around 1% of the patients [2, 3], limits its use to treatment refractory cases of schizophrenia under constant surveillance of the blood cell count.
Clozapine has a complex pharmacological profile interacting with a variety of neurotransmitter receptors in the brain. The exact mechanism of action remains unknown but is assumed to be a combination of dopaminergic and serotonergic effects. According to Burns [2], clozapine has moderate receptor affinity to D1, D2 and D4 receptors, high affinity to H1, α1 and α2, 5-HT2A und 5-HT1C receptors and marked affinity to M1 receptors. Common adverse effects like sedation, hypersalivation and tachycardia are ascribed to the cholinergic, adrenergic as well as histaminergic antagonism.
After administration, clozapine undergoes presystemic elimination and is extensively metabolized by CYP1A2 in the liver [2]. CYP2C19 and CYP3A4 contribute to a smaller extent [4]. The major biotransformation products are norclozapine (N-desmethylclozapine), which is still pharmacologically active, and clozapine-N-oxide. Both can be found in plasma as well as in urine. The structure of the parent compound and the two metabolites are presented in Fig. 1.

Structures of clozapine and its metabolites
Because of its large variability in pharmacokinetic parameters (e.g. oral bioavailability, volume of distribution and half-life therapeutic [3]), the influence of various CYP450 isoenzymes on the metabolism and the wide range for the optimal individual dosing, clozapine is a frequent target for therapeutic drug monitoring.
The daily dose is titrated based on symptom improvement [5]. Usually, therapeutic plasma concentrations in adult patients presenting an acceptable compromise between a good antischizophrenic effect and minimal adverse effects are between 350 and 600 ng/mL and can rarely climb up to 800 ng/mL [6]. Children are more sensitive to clozapine [2], possibly because they produce a greater amount of the active metabolite norclozapine [7]. Consequently, therapeutic plasma concentrations are lower and are found in a range of 200–400 ng/mL [6].
In intoxication cases, clozapine plasma concentrations can increase to microgram-per-milliliter levels. Several clozapine intoxications in children and adults, non-fatal and fatal, accidental or intentional, have been described in the literature [8–12].
Since the time clozapine was established in therapy of schizophrenia several analytical methods using HPLC-UV [13–16], HPLC-DAD [17], CE-DAD [18], GC-MS [19], LC-MS [20–22], and LC-MS/MS [23] have been published. Most of the cited methods also include norclozapine and/or clozapine-N-oxide.
Apart from that, in forensic cases, it can be necessary to detect very low concentration levels in order to prove the intake or application of clozapine—even after several days and in alternative matrices. As urine is easily available and a common specimen in forensic cases, this matrix was included.
The development of this assay was motivated by an intoxication of a 13-month-old girl who was hospitalized after showing respiratory insufficiency, tachycardia and sopor alternating with agitation. We finally received serum and urine samples taken at the (assumed) day of clozapine intake and after 1, 3, 5 and 11 days as well as a cerebro-spinal fluid sample taken at the first day of hospitalization.
Experimental
Chemicals and reagents
Methanolic solutions of clozapine and d4-clozapine were purchased from Lipomed (Bad Säckingen, Germany) and LGC Standards (Wesel, Germany), respectively. Solid powder reference standards of the metabolites norclozapine and clozapine-N-oxide were obtained from Santa Cruz Biotechnology (Heidelberg, Germany).
Methanol (HPLC-grade) was obtained from J.T. Baker (Deventer, The Netherlands), sodium carbonate from Merck (Darmstadt, Germany) and formic acid from Roth (Karlsruhe, Germany). Ethyl acetate, sodium chloride and ammonium formate were purchased from Sigma Aldrich (Steinheim, Germany). All solvents and salts were of analytical grade.
Stock solutions, calibration standards, blank serum and urine
Stock solutions (1.0 mg/mL) of the solid reference standards were prepared using methanol as solvent and stored at −20 °C as all other reference standard solutions.
All stock solutions were then diluted with methanol to obtain appropriate working concentrations (100 μg/mL, 10 μg/mL, 1 μg/mL and 100 ng/mL).
A mixture of the clozapine, norclozapine, and clozapine-N-oxide in methanol was prepared at the same concentration levels as the working solutions to facilitate the spiking of calibration standards later.
The internal standard solution mix contained 5 μg/mL of d4-clozapine in methanol. Drug-free volunteers donated blank serum and blank urine.
Liquid–liquid extraction procedure
For extraction of serum samples, 20 μl of internal standard solution were spiked to an aliquot of 0.5 mL serum (patient samples, QCs or calibrators). One milliliter of an aqueous solution of sodium carbonate (100 g/L, pH ∼11) was added to set alkaline conditions. The serum was then extracted with 1.5 mL ethyl acetate (5 min on a bench-top shaker). After centrifugation for 10 min, an aliquot of the organic layer was transferred into a 1.5-mL vial and evaporated to dryness at 30 °C under reduced pressure (5 mbar) in a vacuum centrifuge (Speedvac, Christ, Osterode, Germany). When dry, the residue was redissolved in 100 μL mobile phase (1 mM ammonium formate/methanol, 80:20, v/v). Twenty microliters was finally injected in the LC-MS/MS system.
The extraction procedure for urine samples was carried out in the same manner except for the sample volume (1.0 mL) and the sodium carbonate solution volume (0.5 mL).
Instrumentation
The Agilent 1100 Series HPLC system consisted of a G1312A binary pump including a G1313A autosampler, a G1379A degasser and a G1316A column oven (Santa Clara CA, USA).
The chromatographic column was a Synergi Polar RP column (150 mm × 2 mm internal diameter, 4 μm, Phenomenex) with a corresponding guard column (Polar RP 4 mm × 2.0 mm) which was kept on a constant temperature of 40 °C.
As a detector, an API 2000 QTrap triple quadrupole linear ion trap mass spectrometer equipped with an atmospheric pressure electrospray ionization interface (TurboIonSpray) was used (Applied Biosystems/MSD Sciex). It was run with Analyst software (version 1.4.2).
LC-MS/MS method
The compounds were separated by gradient elution using 1 mM ammonium formate as mobile phase A and methanol as mobile phase B. The flow rate was 0.4 mL/min.
The gradient was as follows—0 min: 20% organic phase B; 1 min: 20% B; 10 min: 95% B which is held for 3 min; 13 till 13.5 min: decrease from 95% B down to 20%; reequilibration for 2 min.
Data acquisition was performed in MRM mode with positive electrospray ionization.
The ESI inlet conditions were as follows: curtain gas, nitrogen (20 psi); collision gas, nitrogen (5 psi); ion spray voltage, 5,000 V; ion source temperature, 400 °C; ion source gas 1, nitrogen at 40 psi and ion source gas 2, nitrogen at 70 psi.
Declustering, entrance, collision cell entrance and exit potential as well as the collision energy were optimized for the individual transitions; exact values are shown in Table 1.
Validation
-
Selectivity and specificity
In selectivity experiments, six serum and six urine samples from different volunteers were checked for interfering peaks in MRM transitions of analytes as well as of internal standards. Furthermore, two zero samples of each matrix spiked with internal standard solution were tested for undeuterated traces of deuterated clozapine. Additionally, drugs that are sometimes co-administered with clozapine can interfere with the targets. According to the literature, these are benzodiazepines, fluvoxamine, lamotrigin and valproic acid [24, 25]. Samples were spiked with reference standards of these substances at therapeutic levels: fluvoxamine 250 ng/mL, valproic acid 100 μg/mL and lamotrigin 15 μg/mL. A benzodiazepine mixture including nordazepam, diazepam, oxazepam, temazepam, zolpidem, bromazepam, clonazepam, midazolam, flunitrazepam and chlordiazepoxide was added to reach a concentration of 2 μg/mL (0.2 μg/mL for flunitrazepam).
-
Autosampler stability
A common routine run does not exceed 20 h. According to the validation guide of the German Society of Toxicological and Forensic Chemistry (GTFCh) [26] we prepared six quality controls at a low (5 ng/mL) and a high (500 ng/mL) concentration level, extracted them as described above, pooled the final extracts, redivided them in six parts and injected the aliquots in regular time intervals (4 h) into the LC-MS/MS system. The accepted decrease of absolute peak areas is set at 15%.
Storage stability and freeze–thaw stability of clozapine and its metabolite have already been demonstrated by Guitton et al. [14] and Kratzsch et al. [20]. Since all analytes proved to be stable for 1 month (stored at −20 °C) and after three freeze–thaw cycles, we did not need to investigate stability once again.
-
Lower limits of quantification (LOQ)
According to the validation guide of the GTFCh [26] six blank serum and urine samples were spiked with low concentrations of analytes (1.0 ng/mL in serum and 2.0 ng/mL in urine). The analyte concentrations were quantified using a calibration curve whose lowest point was a calibrator with 1 and 2 ng/mL, respectively. Bias and precision of the six measurements were calculated. If they are equal or smaller than 20%, the spiked concentration can be regarded as limit of quantification.
-
Linearity
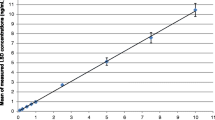
The expected analyte concentrations cover a wide range. Consequently, closely set calibrators were used to generate the calibration curves in serum and urine: 1.0, 2.5, 5.0, 10, 25, 50, 100, 250, 500, 750, 1,000, 1,500 and 2,000 ng/mL. Depending on the concentration of the analyte, the calibration curve was modified by cutting it down to an appropriate low or high concentration range in which linearity could be demonstrated. As described in Peters et al. [27], duplicates at each concentration level were analyzed. The calibration can be considered linear if the calculated concentrations do not pass a range of ±15% of the reference value.
-
Matrix effects, extraction efficencies and recoveries
As proposed by Matuszewski et al. [28], extraction efficiencies and matrix effects were calculated using three different sets of samples. Low-level samples were spiked at 5 ng/mL, high-level samples at 500 ng/mL clozapine, norclozapine and clozapine-N-oxide.
The samples of Set 1 consisted of neat standards containing the analytes in mobile phase. For preparation of Set 2, blank serum and urine samples from five volunteers (0.5 and 1.0 mL, respectively)—plus methanol to imitate the spiking procedure—were extracted by the liquid–liquid extraction method mentioned above. After that, the supernatant was spiked with the analyte mixture and 100 ng d4-clozapine. After evaporation, the residue was reconstituted in 100 μl mobile phase.
Samples of Set 3 were prepared like usual quality control samples. Blank serum and urine samples from the same five volunteers as for Set 2 were spiked with the same amounts of analyte mixture and internal standard. Liquid–liquid extraction followed. The supernatant was then evaporated to dryness and redissolved in mobile phase.
-
Precision and bias
To assess the interday precision and bias pools of serum and urine samples at three concentration levels were prepared, aliquoted and stored frozen at −20 °C. Duplicates were analyzed on six different days. For the low-level quality controls, the concentration was set at 1 (serum) or 2 ng/mL (urine) and for the middle level at 500 ng/mL. Quality controls with a concentration of 5,000 ng/mL (far beyond the upper limit of the calibration range) were used to check whether dilution has an impact on the correctness. Only a fifth of the sample volume was used, filled up with sodium carbonate solution and thenceforth extracted as mentioned above.
Precision was calculated as the relative standard deviation of the replicate analysis, accuracy is the bias in percentage (%) between nominal and measured values. According to widely accepted international guidelines, interday precision and bias in % should be <15% (<20% near the LOQ).
Results and discussion
Chromatography and mass spectrometry
An LC-MS/MS method for detection and quantification of clozapine and its two main metabolites norclozapine and clozapine-N-oxide has been developed.
In the beginning, enhanced product ion spectra in positive ionization mode were monitored as described in [29]. All spectra are depicted in Fig. 2. Table 1 shows the two most abundant fragmentations which were finally selected for MRM transitions, all MS parameters optimized to enhance the performance and the relative ion intensities.

Enhanced product ion spectra of clozapine, norclozapine, clozapine-N-oxide and the internal standard d4-clozapine (collision energies: 20, 35 and 50 eV)
As mentioned by Lin et al. [30] clozapine-N-oxide is thermally unstable and decomposes in gas chromatographic analysis yielding clozapine. Aravagiri et al. [23] therefore kept the ion source temperature at 300 °C in their published LC-MS/MS method. We tested pure clozapine-N-oxide in mobile phase at the proposed ion source temperature and the one that is usually used and could detect only a small difference between spraying at 300 and 400 °C (clozapine peak area in % of clozapine-N-oxide peak area: 1.3% versus 1.9%). As the duration of exposure to elevated temperatures during the electrospray ionization process is much shorter than in a gas chromatographic run, the amount of clozapine generated by reduction of clozapine-N-oxide remains relatively small. It does not affect the quantification neither of clozapine (because of different retention times) nor of clozapine-N-oxide.
The chromatographic separation was achieved in a 15-min run. Norclozapine is the first substance eluting from the column (7.9 min), followed by clozapine (8.4 min) and clozapine-N-oxide (8.9 min). The internal standard d4-clozapine that is the reference parameter for clozapine itself as well as for the two metabolites slightly elutes before clozapine (Fig. 3).
The presented method uses d4-clozapine as deuterated internal standard, monitors two MRM transitions per analyte, works without halogenated solvents and establishes comparable [16] or lower limits of quantification than have been reported in other published methods [12–14, 17, 18, 20–23, 31, 32]. Furthermore, the validation of the presented LC-MS/MS method also includes the assessment of matrix effects in serum and urine which were not given in previous publications.
Validation
-
▪Selectivity
The method was found to be selective for all tested analytes. Selectivity experiments revealed neither interferences of matrix compounds, metabolites, common co-administered drugs or impurities nor undeuterated traces in the deuterated internal standard.
-
▪Autosampler stability
In extracts, all analytes are stable at room temperature over at least 20 h.
-
▪Linearity
Different calibration ranges for quantification of low, middle and high concentrations were established (Table 2) in order to ensure good linearity. They all fulfilled the criterion mentioned above—the calculated concentrations of all calibrators were within the required interval of the reference value ±15%.
-
▪Analytical limits
The limit of quantification was 1.0 ng/mL for all analytes in serum and 2.0 ng/mL for all analytes in urine.
-
▪Extraction efficiencies and matrix effects
The liquid–liquid extraction procedure provided very good results for the extraction efficiencies of clozapine and norclozapine which were 80% for both in serum and 85% and 76% in urine, respectively. Clozapine-N-oxide could be extracted with an efficiency of 52% from serum and of 59% from urine. All results of extraction efficiencies of low and high concentration samples can be found in detail in Table 2.
After extraction from serum, little ion suppression could be observed: matrix effect for norclozapine 77%, for clozapine 85% and for clozapine-N-oxide 87%, in contrast to the fact that matrix effects are generally higher when the analytes are extracted from urine. The highest matrix effect in urine was observed for norclozapine whose signal was suppressed down to 21% of the original pure reference standard signal with a relative standard deviation of 36%. Due to this large variability between the samples norclozapine determination in urine has to be regarded as semi-quantitative. The ion suppression for clozapine and clozapine-N-oxide was 60% and 43% (low concentration samples) while deviating only by 13.2% and 12.9%, respectively.
-
▪Assay precision and bias
For the low and the middle concentrations, the inter-day variations of the spiked quality control samples were <10% for all analytes at both concentration levels in serum as well as in urine. Only the results of norclozapine in urine showed higher deviations: at a concentration of 500 ng/mL, the relative standard deviation was 11.2%; at 2 ng/mL, it was 15.7% but still remained below 20%, which is the enlarged acceptance limit for concentrations near the LOQ.

MRM chromatogram of a spiked serum (a) and urine sample (b) at a concentration level of 10 ng/mL
Bias values were between −9.3% (2 ng/mL norclozapine in urine) and 8.5% (clozapine-N-oxide in urine) and easily fulfilled the established limits.
Diluting samples and deducing their results is possible for norclozapine, clozapine (in both matrices) and clozapine-N-oxide in serum. Precision values ranged from 4.8% to 7.45% and bias values from 1.9% to 15.0%. In contrast, dilution severely affects the quantification of clozapine-N-oxide in urine and leads to overestimation of the concentrations. As a consequence, an additional calibration with diluted calibrators has to be established for this analyte if needed. Otherwise, the calibration has to be extended up to higher concentration levels.
Detailed results of all validation parameters are given in Table 2.
Samples of the intoxication case
All remaining samples that had been taken from the intoxicated child during its stay in hospital regardless of matrix, amount and state were collected for later analysis. Finally, five serum, three urine and one cerebro-spinal fluid sample could be extracted and analysed. If the available sample amount did not reach the sample volume used for calibration, it was filled up with sodium carbonate solution.
Cerebro-spinal fluid of healthy humans is an aqueous fluid with a pH of 7.8 containing nearly no protein and no cells. Its composition can change in cases of bleeding, infection, autoinflammation or a dysfunction of metabolism but the analysed cerebro-spinal fluid sample did not show any abnormalities (2 cells/μl, 18 mg/dL protein). Lacking blank cerebro-spinal fluid for validation experiments and calibrations, we used isotonic solution of sodium chloride as matrix surrogate. One calibration curve was assumed to be sufficient to prove linearity and to calculate the analyte concentrations in the single cerebro-spinal fluid sample. No further assessment of validation parameters was performed as the analysis of cerebro-spinal fluid is uncommon in forensic toxicology and of yet unknown clinical impact.
Whereas in the beginning, analyte concentrations were assumed to be very high, in the end they dropped down to reach the limit of quantification or even lower. Clozapine concentrations in serum were in the range of 18 to 736 ng/mL, in the urine sample of the first day we found 193 ng/mL. Visible but not quantifiable clozapine traces could still be detected in the serum and urine sample taken at the fifth day after intake. As only 40 μl of the 5-day-serum sample were left for the analysis, the clozapine trace peak was calculated by extrapolation and resulted in a clozapine concentration of around 1 ng/mL.
Norclozapine ranged from 2.9 to 300 ng/mL in serum and from 17.7 to 1,730 ng/mL in urine. In the last urine sample, a trace amount that yielded a concentration below the limit of quantification was visible.
Clozapine-N-oxide concentrations ranged from 2.0 to 174 ng/mL in serum and from 0.11 to 5,500 ng/mL in urine. The urine sample containing the high concentration of clozapine-N-oxide was diluted (1:5) prior to analysis. An extra calibration with 1:5 diluted urine samples served for quantification.
Cerebro-spinal fluid concentrations were as follows: norclozapine 2.0 ng/mL, clozapine 3.6 ng/mL and clozapine-N-oxide 1.3 ng/mL.
Conclusion
The extraction and analysis procedure reported here presents a selective, sensitive and accurate method for the simultaneous quantification of clozapine and its metabolite clozapine-N-oxide in serum and urine and for norclozapine in serum. It allows for the fast determination of concentrations in the low nanogram-per-milliliter range. The application of the method in a clinical intoxication case confirmed its suitability for forensic and clinical purposes.
References
Luft B, Taylor D (2006) A review of atypical antipsychotic drugs versus conventional medication in schizophrenia. Expert Opin Pharmacother 7:1739–1748
Burns MJ (2001) The pharmacology and toxicology of atypical antipsychotic agents. J Toxicol Clin Toxicol 39:1–14
Dubois D (2005) Toxicology and overdose of atypical antipsychotic medications in children: does newer necessarily mean safer? Curr Opin Pediatr 17:227–233
Jaquenoud Sirot E, Knezevic B, Morena GP, Harenberg S, Oneda B, Sv C, Ansermot N, Baumann P, Eap CB (2009) ABCB1 and cytochrome P450 polymorphisms: clinical pharmacogenetics of clozapine. J Clin Psychopharmacol 29:319–326. doi:10.1097/JCP.0b013e3181acc372
Gogtay N, Rapoport J (2008) Clozapine use in children and adolescents. Expert Opin Pharmacother 9:459–465
The International Association of Forensic Toxicologists (2010) www.tiaft.org
Frazier JA, Cohen LG, Jacobsen L, Grothe D, Flood J, Baldessarini RJ, Piscitelli S, Kim GS, Rapoport JL (2003) Clozapine pharmacokinetics in children and adolescents with childhood-onset schizophrenia. J Clin Psychopharmacol 23:87–91
Ishii A, Mizoguchi K, Kageoka M, Seno H, Kumazawa T, Suzuki O (1997) Nonfatal suicidal intoxication by clozapine. J Toxicol Clin Toxicol 35:195–197
Keller T, Miki A, Binda S, Dirnhofer R (1997) Fatal overdose of clozapine. Forensic Sci Int 86:119–125
Klys M, Rojek S, Rzepecka-Wozniak E (2007) Neonatal death following clozapine self-poisoning in late pregnancy: an unusual case report. Forensic Sci Int 171:e5–e10
Mady S, Wax P, Wang D, Goetz C, Hadley C, Love R (1996) Pediatric clozapine intoxication. Am J Emerg Med 14:462–463
Renwick AC, Renwick AG, Flanagan RJ, Ferner RE (2000) Monitoring of clozapine and norclozapine plasma concentration-time curves in acute overdose. J Toxicol Clin Toxicol 38:325–328
Avenoso A, Facciola G, Campo GM, Fazio A, Spina E (1998) Determination of clozapine, desmethylclozapine and clozapine N-oxide in human plasma by reversed-phase high-performance liquid chromatography with ultraviolet detection. J Chromatogr B Biomed Sci Appl 714:299–308
Guitton C, Kinowski JM, Aznar R, Bressolle F (1997) Determination of clozapine and its major metabolites in human plasma and red blood cells by high-performance liquid chromatography with ultraviolet absorbance detection. J Chromatogr B Biomed Sci Appl 690:211–222
Mercolini L, Bugamelli F, Kenndler E, Boncompagni G, Franchini L, Raggi MA (2007) Simultaneous determination of the antipsychotic drugs levomepromazine and clozapine and their main metabolites in human plasma by a HPLC-UV method with solid-phase extraction. J Chromatogr B Analyt Technol Biomed Life Sci 846:273–280
Shen YL, Wu HL, Ko WK, Wu SM (2002) Simultaneous determination of clozapine, clozapine N-oxide, N-desmethylclozapine, risperidone, and 9-hydroxyrisperidone in plasma by high performance liquid chromatography with ultraviolet detection. Anal Chim Acta 460:201
Titier K, Bouchet S, Pehourcq F, Moore N, Molimard M (2003) High-performance liquid chromatographic method with diode array detection to identify and quantify atypical antipsychotics and haloperidol in plasma after overdose. J Chromatogr B Analyt Technol Biomed Life Sci 788:179–185
Raggi MA, Bugamelli F, Mandrioli R, Sabbioni C, Volterra V, Fanali S (2001) Rapid capillary electrophoretic method for the determination of clozapine and desmethylclozapine in human plasma. J Chromatogr A 916:289–296
Markowitz JS, Patrick KS (1995) Thermal degradation of clozapine-N-oxide to clozapine during gas chromatographic analysis. J Chromatogr B Biomed Appl 668:171–174
Kratzsch C, Peters FT, Kraemer T, Weber AA, Maurer HH (2003) Screening, library-assisted identification and validated quantification of fifteen neuroleptics and three of their metabolites in plasma by liquid chromatography/mass spectrometry with atmospheric pressure chemical ionization. J Mass Spectrom 38:283–295
Niederlander HA, Koster EH, Hilhorst MJ, Metting HJ, Eilders M, Ooms B, de Jong GJ (2006) High throughput therapeutic drug monitoring of clozapine and metabolites in serum by on-line coupling of solid phase extraction with liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 834:98–107
Rao LV, Snyder ML, Vallaro GM (2009) Rapid liquid chromatography/tandem mass spectrometer (LCMS) method for clozapine and its metabolite N-desmethyl clozapine (norclozapine) in human serum. J Clin Lab Anal 23:394–398
Aravagiri M, Marder SR (2001) Simultaneous determination of clozapine and its N-desmethyl and N-oxide metabolites in plasma by liquid chromatography/electrospray tandem mass spectrometry and its application to plasma level monitoring in schizophrenic patients. J Pharm Biomed Anal 26:301–311
Deutsche Gesellschaft für Psychiatrie DGPPN (2005) Leitlinie Psychiatrie—Schizophrenie. http://www.dgppn.de/publikationen/leitlinien/leitlinien0.html
Lu ML, Lane HY, Chen KP, Jann MW, Su MH, Chang WH (2000) Fluvoxamine reduces the clozapine dosage needed in refractory schizophrenic patients. J Clin Psychiatry 61:594–599
Peters FT, Hartung M, Herbold M, Schmitt G, Daldrup T, Musshoff F, Paul L, Aebi B, Auwaerter V, Kraemer T, and Skopp G (2009) Richtlinien der GTFCh zur Qualitätssicherung bei forensich-toxikologischen Untersuchungen, Anhang B: Anforderung and die Validierung von Analysenmethoden
Peters FT, Drummer OH, Musshoff F (2007) Validation of new methods. Forensic Sci Int 165:216–224
Matuszewski BK, Constanzer ML, Chavez-Eng CM (2003) Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem 75:3019–3030
Dresen S, Gergov M, Politi L, Halter C, Weinmann W (2009) ESI-MS/MS library of 1,253 compounds for application in forensic and clinical toxicology. Anal Bioanal Chem 395:2521–2526
Lin G, McKay G, Hubbard JW, Midha KK (1994) Decomposition of clozapine N-oxide in the qualitative and quantitative analysis of clozapine and its metabolites. J Pharm Sci 83:1412–1417
Ho YH, Ko WK, Kou HS, Wu HL, Wu SM (2004) Field-amplified sample stacking in capillary electrophoresis for the determination of clozapine, clozapine N-oxide, and desmethylclozapine in schizophrenics' plasma. J Chromatogr B Analyt Technol Biomed Life Sci 809:111–116
Mercolini L, Grillo M, Bartoletti C, Boncompagni G, Raggi MA (2007) Simultaneous analysis of classical neuroleptics, atypical antipsychotics and their metabolites in human plasma. Anal Bioanal Chem 388:235–243
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wohlfarth, A., Toepfner, N., Hermanns-Clausen, M. et al. Sensitive quantification of clozapine and its main metabolites norclozapine and clozapine-N-oxide in serum and urine using LC-MS/MS after simple liquid–liquid extraction work-up. Anal Bioanal Chem 400, 737–746 (2011). https://doi.org/10.1007/s00216-011-4831-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-4831-8