
Abstract
A rapid multi-analyte method has been developed for the simultaneous determination of pesticides and mycotoxins in milk by ultra high-performance liquid chromatography coupled to triple quadrupole mass spectrometry (UHPLC–QqQ–MS/MS). A variety of methodologies has been evaluated, including solid-phase extraction (SPE), “dilute-and-shoot” (liquid–liquid extraction-based procedures), and QuEChERS (quick, easy, cheap, effective, rugged, and safe)-based methods. The optimization and development process was carried out considering that the maximum residue level for aflatoxin M1 (AFM1) in milk in the European Union (EU) is set at 0.05 μg kg−1, which is the lowest tolerance in the target compounds. The selected method consisted of an extraction by SPE using C18 as sorbent and methanol as elution solvent. The final determination was performed by UHPLC–QqQ–MS/MS. Matrix-matched standard calibration was used for quantification, obtaining recoveries in the range 60–120% with relative standard deviations <25%, at three spiking levels: 0.5, 10, and 50 μg kg−1 (ten times lower for AFM1). Limits of quantification ranged from 0.20 to 0.67 μg kg−1, which were always below or equal to the established tolerance levels by the EU. Finally, the selected method was applied to different types of milk.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Milk and dairy products are essential in the human diet due to their important nutritive qualities (i.e., they represent the main source of calcium necessary for the human growth) [1]. However, the consumption of these products is not free from the risk of exposure to harmful compounds, such as pesticides and mycotoxins. It is well known that pesticides are synthetic products used for the management of pests in agricultural production and for the control of animal parasites, whereas mycotoxins are toxic natural secondary metabolites generated under special environmental conditions by the metabolism of certain fungus, such as Fusarium, Aspergillus flavus, Aspergillus nomius, and Aspergillus parasiticus [2]. These organisms can grow in food products (especially cereal crops) as a consequence of storage and/or food processing. Therefore, they can reach the food chain through the consumption of contaminated fodder, pastures, or feed by livestock [3, 4]. The presence of mycotoxins and pesticides may constitute a serious problem for human health, due to the high toxicity and harmful effects to the human system [5, 6], such as the carcinogenic effect observed for some of them [2, 7].
Among all mycotoxins that can appear in animal feed, aflatoxins are the most relevant compounds because they are most likely to be found in milk. Within this group, the main concern in milk is aflatoxin M1 (AFM1), which is the major metabolite of aflatoxin B1 (AFB1). This mycotoxin is poorly degraded in rumen microorganisms, and it is mainly metabolized in the liver into AFM1 [2], which shows resistance to the pasteurization process, and therefore, it can appear in ultra high temperature (UHT) milk [7]. In spite of AFM1 having the highest probability to be detected in milk, other mycotoxins can be transferred to milk, such as HT-2 toxin, T-2 toxin, and ochratoxin A [8–10].
Consequently, in order to ensure milk safety, the US Food and Drug Administration (FDA) and the European Union (EU) have established maximum residue levels (MRLs) for pesticides [11] and AFM1 [12, 13], although for other mycotoxins, such as some aflatoxins (G1, G2, B1, and B2), MRLs have not been set in this food commodity. Furthermore, it is important to notice that the MRL established by the EU for AFM1 (0.05 μg kg−1) is extremely low, and it is further below the tolerance set by the FDA (0.50 μg kg−1).
In order to allow an extensive and effective control of the occurrence of pesticides and mycotoxins in milk, a number of sensitive and reliable methodologies have been developed for the analysis of each group of contaminants separately. One of the main difficulties related to the determination of these analytes in milk is its high fat and protein content that can often interfere in the analytical determination. For this reason, sample extraction can result long and tedious, involving several clean-up steps to remove the co-extracted material from the matrix.
The methods described in literature for the extraction of mycotoxins from milk utilized different extraction techniques, such as liquid–liquid extraction (LLE) [9], solid-phase extraction (SPE) [14, 15], or LLE followed by a SPE clean-up step [10]. The use of SPE with immunoaffinity sorbents has often been used in mycotoxin extraction [8, 16] and for clean-up purposes [8, 17]. These extraction methods normally include a pre-concentration step due to the low MRLs for mycotoxins and especially for AFM1 in milk.
Although conventional techniques, such as SPE [5, 18] and LLE [19], have also been described for the extraction of pesticides from milk, several approaches applying other techniques have been evaluated, such as matrix solid-phase dispersion (MSPD) [20], dispersive solid-phase extraction (D-SPE) [21], liquid-phase microextraction (LPME) [22], and solid-phase microextraction (SPME) [23]. Among all procedures employed for the extraction of pesticides, the QuEChERS method (acronymic name from quick, easy, cheap, effective, rugged, and safe) [24], which was originally developed for the extraction of pesticides from fruits and vegetables, has been employed in many other matrices, such as olive oil or fruit juices [25, 26]. Nevertheless, it has not been employed in the extraction of pesticides from milk so far.
As a general rule, the control of these two groups of undesirable substances in milk would involve the performance of two different analyses for each group, requiring extra efforts, causing an increase in the analysis time and affecting sample-throughput. In this sense, there is a current trend for the development of multi-analyte methods able to analyze several classes of compounds simultaneously. Obviously, the increase in the scope of the methods benefits to the sample throughput since a high number of compounds can be determined in a single analysis. Nevertheless, the physicochemical properties of the compounds included in each group of contaminants can have very different results, hindering the development of single adequate extraction procedures. In the area of mycotoxins and pesticides, only two papers have been focused on a simultaneous determination in food. Lacina et al. [27] developed a method for the analysis of pesticides and mycotoxins in cereals (22 mycotoxins and 222 pesticides were evaluated), and Mol et al. [28] developed a generic extraction method for pesticides, natural toxins, and veterinary drugs in different food matrices, including milk (20 mycotoxins and 172 pesticides were evaluated). Despite of the high number of compounds covered by this last method, AFM1 was not included in the study.
With regard to the separation and detection technique, liquid chromatography coupled to tandem mass spectrometry detection (LC–MS/MS) is widely applied for the determination of mycotoxins and pesticides. Besides, ultra high-performance liquid chromatography (UHPLC) presents several benefits in the development of multi-analyte analysis, such as reduction of the running time or improvement in sensitivity and peak resolution. Some applications of this technique have been reported for determination of mycotoxins [29, 30] and pesticides [26, 31] in the food field, but its application in generic determinations is still very scarce [28].
The aim of this work is the combination of an easy extraction methodology with appropriate LC conditions in order to have a simple and fast method for the simultaneous analysis of mycotoxins and pesticide residues in milk. To achieve this goal, several multi-residue or multi-analyte methods have been tested using UHPLC coupled to triple quadrupole MS/MS (UHPLC–QqQ–MS/MS) for the final determination. The final optimized method has been validated in order to ensure the adequate analysis of AFM1 in milk together with the other target compounds.
Materials and methods
Reagents and chemicals
Aflatoxins B1, B2, G1, and G2 pure standards and stock solutions of AFM1 and HT-2 toxin (in acetonitrile) were purchased from Sigma-Aldrich (Madrid, Spain). Ochratoxin A and T-2 toxin were obtained from Biopure (Tulln, Austria). Pesticide standards (purity > 99%) were purchased from Dr. Ehrenstofer (Augsburg, Germany) and Riedel-de-Haën (Seelze-Hannover, Germany). Individual stock solutions of 200 mg L−1 of all standards were prepared by exact weighing of powder or liquid and dissolved in 50 mL of HPLC-grade acetonitrile for mycotoxins (J.T. Baker, Deventer, Holland) or HPLC-grade methanol for pesticides (J.T. Baker). Two working standard solutions (2 mg L−1) were prepared from the stock solutions in acetonitrile (mycotoxins) and methanol (pesticides). A final working standard solution of pesticides and mycotoxins at a concentration of 1 mg L−1 (except for AFM1, which was 0.1 mg L−1) was prepared from the aforementioned working standard solutions.
Ammonium formate was obtained from Panreac (Barcelona, Spain). Ultrapure water was obtained from a Milli-Q gradient water system (Millipore, Bedford, MA, USA). Ethyl acetate, n-hexane, and dichlorometane were purchased from J.T. Baker, and acetone was obtained from Fluka (Seelze, Germany). Oasis HLB SPE (200 mg/6 cm3) and C18 Sep-Pak (200 mg/3 cm3) cartridges were purchased from Waters (Milford, MA, USA), while BondElut Jr florisil cartridges (500 mg) were provided by Varian (Harbor City, CA, USA). These cartridges were used in clean-up stages and SPE procedures. Anhydrous magnesium sulfate, sodium chloride, and sodium acetate were purchased from Panreac. Sodium citrate dibasic sesquihydrate was obtained from Sigma-Aldrich, and sodium citrate dihydrate was obtained from J.T. Baker.
A vortex mixer (model Reax 2000), a rotary agitator (model Reax-2, end-over-end) from Heidolph (Schwabach, Germany), and an analytical AB204-S balance (Mettler Toledo, Greinfesee, Switzerland) were also used. Millex-GN nylon filters (0.20 μm, Millipore, Carrightwohill, Ireland) were used for filtration of sample extracts. An extraction manifold from Waters connected to a Büchi Vac V-500 (Flawil, Switzerland) vacuum system was used for SPE experiments.
UHPLC–QqQ–MS/MS analysis
Chromatographic analyses were performed using an Acquity UPLC™ system, and separations were carried out using an Acquity UPLC™ BEH C18 column (100 × 2.1 mm, 1.7 μm particle size) from Waters. The C18 column was equilibrated at 30 °C. The analytes were separated with a mobile phase consisting of methanol (eluent A) and an aqueous solution of ammonium formate 5 mM (eluent B) at a flow rate of 0.35 mL min−1. The analysis started with 25% of eluent A, which was increased linearly up to 100% in 3.75 min; this composition was held for 1.25 min before being returned to 25% of eluent A in 0.5 min, followed by a re-equilibration time of 1 min (total run time, 6.5 min). The injection volume was always 5 μL.
MS/MS analysis was carried out using a Waters Acquity TQD tandem quadrupole mass spectrometer (Waters, Manchester, UK). The instrument was operated using an electrospray source in positive mode (ESI (+)). ESI parameters were as follows: capillary voltage, 3.5 kV; extractor voltage, 3 V; source temperature, 120 °C; desolvation temperature, 350 °C; cone gas (N2) flow, 80 L h−1; and desolvation gas (N2) flow, 600 L h−1. Collision-induced dissociation was performed using argon as the collision gas at a pressure of 4 × 10−3 mbar in the collision cell. The specific MS/MS parameters for each compound are shown in Table 1. Data acquisition was performed using MassLynx 4.0 software with QuanLynx program (Waters).
Sample preparation
Method A: QuEChERS method using citrate buffer
Ten milliliters of milk was weighed into a 50-mL polypropylene centrifuge tube, and 10 mL of acetonitrile (1% acetic acid, v/v) was added [32]. The mixture was shaken in the vortex for 1 min. Then, 4 g of anhydrous magnesium sulfate, 1 g of sodium chloride, 1 g of sodium citrate dehydrate, and 0.5 g of disodium hydrogencitrate sesquihydrate were added. The tube was shaken vigorously for 1 min and centrifuged for 5 min at 5,000 rpm (4,136×g). An aliquot (2 mL) of the acetonitrile layer was filtered through a nylon filter and transferred into an autosampler vial for injection into the UHPLC–QqQ–MS/MS system. Despite the conventional QuEChERS implying a dispersive SPE clean-up step, using primary secondary amine as sorbent material, in this work, this step was avoided.
Method B: QuEChERS method using acetate buffer
This was the same as the previous method A, but with 1 g of sodium acetate instead of 1 g of sodium chloride, 1 g of trisodium citrate dehydrate, and 0.5 g of disodium hydrogencitrate sesquihydrate [33, 34].
Method C: LLE-based method “dilute-and-shoot” (option 1)
Analytes were extracted from milk using an extraction method based on the procedure previously described by Mol et al. [28]. Briefly, 2.5 g of milk was diluted with 5 mL of Milli-Q water, and the mixture was shaken in a vortex. Then, 15 mL of acetonitrile (1% formic acid, v/v) was added, and the sample was put into the rotary agitator, and it was extracted by end-over-end shaking for 1 h at 50 rpm. After that, the mixture was centrifuged for 10 min at 5,000 rpm (4,136×g), and 1 mL of the supernatant was filtered through a nylon filter and transferred into an autosampler vial for injection in the UHPLC–QqQ–MS/MS system.
Method D: LLE-based method “dilute-and-shoot” (option 2)
This was the same as proposed method C but with acetone (1% formic acid, v/v) instead of acetonitrile.
Clean up stage (methods A, B, C, and D)
For the clean-up experiments carried out after the four protocols described above, an aliquot of supernatant (3 mL) was transferred into an SPE cartridge, evaluating two types of sorbents: C18 and Oasis HLB. The cleaned extract was collected, and 1 mL was filtered through a nylon filter and transferred into an autosampler vial for injection in the UHPLC–QqQ–MS/MS system.
Method E: recommended and validated SPE method
Ten millimeters of milk was diluted with 10 mL of Milli-Q water. The sample was centrifuged for 10 min at 5,000 rpm (4,136×g). Then, the supernatant was loaded into a C18 cartridge previously conditioned with 5 mL of methanol and 5 mL of Milli-Q water. Afterwards, the cartridges were washed with 5 mL of water and 5 mL of n-hexane, and they were vacuum-dried for 30 min. The analytes were eluted with methanol (5 mL). The eluate was evaporated under a gentle N2 stream, and the residue was dissolved in 1 mL of mobile phase (methanol and aqueous solution of ammonium formate, 5 mM, 1:4, v/v), prior to the chromatographic analysis.
Validation
Performance characteristics of the optimized method were established by a validation procedure according to the criteria laid down in the European Commission Decision 2002/657/CE [35] and European SANCO guideline [36]. Linearity of the method was evaluated using matrix-matched standard calibration by analyzing spiked blank samples of milk at four concentration levels in the range 0.5–150 μg L−1 for all compounds, except for AFM1 (0.05–15 μg L−1). The concentration for AFM1 in all the validation experiments was ten times lower. In order to evaluate the trueness of the selected method, recovery studies were carried out at three concentration levels (0.5, 10, and 25 μg kg−1) by spiking five blank samples at each level. Repeatability (n = 5) was studied at three concentration levels (0.5, 10, and 25 μg kg−1). Intermediate precision was evaluated by analyzing spiked samples at 25 μg kg−1 (2.5 μg kg−1 for AFM1) in six different days.
The specificity of the method was evaluated by the analysis of a blank sample and a spiked blank sample at 25 μg kg−1 (ten times lower for AFM1). Finally, the limits of detection (LOD) and quantification (LOQ) were calculated analyzing matrix-matched standards at 0.1, 0.2, 0.5, 1, and 2 μg kg−1 (concentrations ten times lower for AFM1). The LODs and LOQs were determined as the lowest concentration of the analyte yielding a signal-to-noise ratio (S/N) of 3 and 10, respectively.
Results and discussion
The aim of this work was the development of a multi-analyte method allowing the simultaneous extraction and analysis of pesticides and mycotoxins in milk. The selected compounds cover a wide range of polarity, including LC-amenable pesticides that must be determined according to the current legislation [37], pesticides whose application is forbidden and the most studied mycotoxins.
Due to the low MRL set by the EU for AFM1 (0.05 μg kg−1 in milk), the optimized methodology must be able of detecting this analyte at this level. Subsequently, during the development of the method, the sensitivity for this compound was an important issue to select the most suitable extraction technique.
UHPLC–QqQ–MS/MS analysis
Two UHPLC–QqQ–MS/MS methods reported in previous studies for the multi-residue determination of pesticides [26] and mycotoxins [29] were considered for the development of common conditions. Both methods utilized methanol as organic solvent because it provided the best sensitivities for all compounds. However, different aqueous phases were applied. The pesticide method used an aqueous solution of formic acid (0.01%, v/v), whereas for mycotoxins, the optimal results were obtained employing an aqueous solution of ammonium formate 5 mM. Bearing in mind that some mycotoxins (i.e., aflatoxins, T-2, and HT-2) showed worse sensitivity when aqueous formic acid was used, the ammonium formate solution was therefore selected for the simultaneous determination. The overall analysis time operating under these conditions was 6.5 min. The retention time ranged from 1.20 (propamocarb) to 4.78 min (fenpropimorph), and analyte retention time variations were ≤0.5%.
For MS/MS determination, ESI (+) was used, and two transitions per compound were monitored. Table 1 shows the specific conditions for each compound. The analytes were distributed in 20 functions, using a maximum of ten compounds per function. Different dwell times were evaluated for each function, and the optimum values ranged from 0.005 to 0.100 s.
Optimization of the extraction method
As previously commented, different procedures have been reported in literature for the extraction of pesticides and mycotoxins from milk, although a limited number of them have carried out a simultaneous determination. In this work, several procedures have been evaluated, bearing in mind the recovery at low concentrations for AFM1 and the number of compounds showing adequate recovery values.
One of the methodologies evaluated was the QuEChERS method due to it is a multi-residue extraction approach widely used in the field of pesticides for the analysis of compounds with different polarity [26]. The experiments were carried out employing spiked blank skimmed milk samples at 50 μg kg−1, except for AFM1 (5 μg kg−1). Two QuEChERS methods were tested: the acetate and the citrate-based QuEChERS methods. Currently, the QuEChERS method using acetate buffer is the official method of the AOAC [24, 33], whereas the citrate-based QuEChERS procedure has been set as the official method by the European Norm [32, 34]. An additional clean-up stage by SPE (C18 and Oasis HLB cartridges) was also evaluated in order to obtain extracts with less co-extracted material (“Sample preparation” section). The best results were obtained with the citrate-based method using a clean-up step by SPE with Oasis HLB (38 compounds; Table 2). However, when this methodology was evaluated with full-cream milk, the AFM1 was not properly extracted, and the number of compounds showing adequate recovery decreased dramatically (8 compounds). The following optimization experiments were carried out using full-cream milk samples as the most complex milk matrix.
Due to the unsatisfactory performance of the QuEChERS-based methodologies, the evaluation of the generic method proposed by Mol et al. [28] was tested. This “dilute-and-shoot” procedure was developed for a wide variety of matrices and compounds, including pesticides and mycotoxins. However, this new approach did not monitor AFM1 in milk. The optimized method involved a dilution with water followed by LLE with a water-miscible organic solvent, obtaining the best overall results using acidified acetonitrile. However, the specific results for milk were better using acidified acetone, which was selected for its evaluation on spiked full-cream milk samples at 250 μg kg−1 (ten times lower for AFM1). Because the extracts obtained after centrifugation were moderately turbid, an SPE clean-up step using Oasis HLB was additionally evaluated. The most adequate results were achieved without any clean-up step (54 compounds; Table 2). However, the extraction method did not provide acceptable recoveries for AFM1 at the MRL level.
Initially, one of the aims of the study was the application of a simple and fast method on the basis of either the QuEChERS or “dilute-and-shoot” strategies. However, these multi-residue and generic methodologies did not permit the extraction of AFM1 at the EU MRL levels, demonstrating that they can fail for difficult compounds showing very low tolerances. For this reason, an alternative SPE method was considered since it would allow the performance of a concentration of the analytes without increasing the matrix content in the final extract. In order to optimize the extraction step by SPE, the type of cartridge and the solvent elution were studied. In literature, Oasis HLB cartridges [10] and C18 bonded silica [15] were utilized for the extraction of 18 mycotoxins and AFM1, respectively. These sorbents were therefore tested. The solvent elution and the washing step were also evaluated. The obtained results are shown in Fig. 1. The C18 cartridges allowed the extraction of a higher number of compounds with adequate recoveries than Oasis HLB, and it was used for further experiments.

Recovery results obtained for the SPE extraction of spiked milk samples (25 μg kg−1, ten times lower for AFM1) using C18 and Oasis sorbents. Pest pesticides, Mycot mycotoxins. Extraction conditions: elution solvent, methanol; no dilution step prior SPE; no washing step. Other conditions were the same as described in “Sample preparation” section
Certain clogging problems were observed in the cartridges, probably because of the high-size molecules (i.e., lipids and proteins) that are present in milk. Samples were therefore centrifuged to remove partially this matrix content. Besides, two simple pre-treatment steps were also considered to reduce the matrix content: protein precipitation by freezing and dilution. The freezing of milk samples was carried out at −25 °C for 2 h, whereas the dilution was performed with water. Both procedures were followed by centrifugation, and the remaining supernatants were then extracted by SPE. The obtained results showed that higher recovery values were achieved when the milk was submitted to dilution (data not shown).
After these preliminary experiments, the elution solvent was evaluated. Several mixtures showing different polarity were tested: dichloromethane:acetone (95:5, v/v) [15], ethyl acetate:acetone (95:5, v/v) mixture, acetone, and methanol. Methanol provided the best results (Fig. 2). Thus, 48 compounds were extracted when methanol was used as elution solvent, 24 compounds with acetone, 21 compounds with dichloromethane:acetone (95:5, v/v), and 17 compounds with ethyl acetate:acetone (95:5, v/v). In relation to AFM1, adequate recoveries were obtained when methanol and the dichloromethane mixture were used; however, methanol provided higher recovery for AFM1, and it was finally selected as the most suitable elution solvent.

Recovery results obtained for the SPE extraction of spiked milk samples (25 μg kg−1, ten times lower for AFM1) using different elution solvents. EtAc ethyl acetate, MeOH methanol. Extraction conditions: cartridge, C18; no washing step. Other conditions were the same as described in “Sample preparation” section
The application of a washing step before elution was studied. Three different conditions were tested, some of them reported in literature [10, 15]: no washing step, 5 mL of water, and 5 mL of water plus 5 mL of n-hexane. The best results were achieved when the cartridges were washed with water and n-hexane (data not shown).
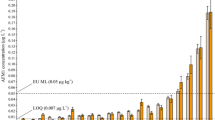
Although the evaluated methods could extract simultaneously mycotoxins and pesticides, SPE was the only one that allows the simultaneous extraction of 42 pesticides and six mycotoxins (including AFM1) with good recovery values (60% < R < 120%) at low concentration levels (Fig. 3). Therefore, these compounds were considered for validation.

UHPLC–QqQ–MS/MS extracted chromatogram of several pesticides and mycotoxins of a spiked milk sample (2 μg kg−1, 0.2 μg kg−1 for aflatoxin M1) analyzed with the validated method
Method validation
Firstly, the matrix effect (suppression/enhancement of the signal in the ionization source) was evaluated by injecting several concentrations (0.5–150 μg L−1, ten times lower for AFM1) in pure solvent and in extracted blank milk samples. The slopes of the calibration curves were compared using the procedure indicated by Cuadros-Rodríguez et al. [38]. It can be observed that the slopes are statistically different (p value lower than 5%) for most of the compounds assayed (Table 3), except for methomyl, epoxiconazole, and tebutam. In consequence, the presence of a matrix effect was demonstrated, and matrix-matched standards (full-cream milk) were used, showing good linearity, with determination coefficients (R 2) higher than 0.9980 for all compounds in the range from 0.5 to 150 μg kg−1 (ten times lower for AFM1).
Trueness was evaluated through recovery studies by spiking blank samples at three concentration levels: 0.5, 10, and 25 μg kg−1 (n = 5). The recoveries obtained by the optimized method (Table 3) were in the range 61.4–118.2% at 0.5 μg kg−1 (except for vamidothion, 125.1%), 60.2–110.4% at 10 μg kg−1 (except for atrazine, iodosulfuron methyl, and tepraloxydim, recoveries higher than 120%), and 60.7–119.2% at 25 μg kg−1 (except for imidacloprid, monolinuron, and atrazine with recoveries values higher than 120%). The method could not be validated at 0.5 μg kg−1 for 2,6-dichlorobenzamide, chlorsulfuron, and thiabendazole. Thiabendazole and chlorsulfuron were determined at concentrations higher than 10 μg kg−1, and 2,4-dichlorobenzamide was determined at concentrations higher than 25 μg kg−1. For AFM1, recoveries were evaluated at 0.05 (84.5%), 1 (71.9%), and 2.5 (97.3%) μg kg−1.
Repeatability was studied at the concentration levels assayed for the recovery studies, performing five replicates for each level. The method showed good repeatability (relative standard deviation (RSD) <20%) for all the analyzed compounds, except for some analytes. For instance, cinosulfuron, carbaryl, azaconazole, linuron, and epoxiconazole showed RSDs values lower than 30% at 0.5 μg kg−1, and acetamiprid, chlorsulfuron, azaconazole, and tebutam presented RSDs lower than 25% at 10 μg kg−1.
Inter-day precision was studied by the analysis of five spiked samples at 25 μg kg−1 (ten times lower for AFM1) in five consecutive days. RSDs lower than 25% were obtained for all compounds except for imidacloprid, cinosulfuron, iodosulfuron methyl, simazine, and tepraloxidim with RSDs lower than 30%. Bearing in mind that acceptable precision and recovery values were obtained, the use of internal standard was not considered.
LODs and LOQs were lower than 0.20 and 0.67 μg kg−1, respectively (Table 3). It must be noticed that the LOQ obtained for all compounds were lower than MRLs. For instance, LOQ for AFM1 was 0.03 μg kg−1, which is a concentration lower than the MRL established for this mycotoxin in milk (0.05 μg kg−1).
Finally, the selectivity of the method was evaluated by analyzing control blank samples. The absence of any signal at the same retention time as the selected compounds indicated that there were not any matrix interferences that may give a false positive signal.
Sample analysis
The validated analytical method was used for the analysis of 15 real samples of milk purchased from several local markets. Different types of milk were selected (three samples of skimmed milk, four samples of semi-skimmed milk, two samples of full-cream milk, three samples of milk enriched with calcium, and three samples of powdered milk-based infant formula). An internal quality control (IQC) was carried out for every batch of samples in order to check if the system was under control. This IQC implied the preparation of a matrix-matched standard calibration, a reagent blank, a matrix blank, and a spiked blank sample at 25 μg kg−1 (ten times lower for AFM1), which were injected together with each batch of samples in order to check the performance of the method.
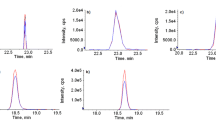
Table 4 shows the results obtained. Some pesticides (acetamiprid, thiacloprid, carbendazim, thiabendazole, thiophanate methyl, and bendiocarb) were detected, whereas mycotoxins were not detected in the analyzed samples. It must be highlighted that imidacloprid and atrazine desisopropyl were detected in four samples at concentration levels higher than 10 μg kg−1; currently, there is not any MRL set for them in milk. The presence of imidacloprid could be explained considering that this pesticide is usually employed as foliar application in certain crops, which can be used for feed production, and to control fleas [39]. Figure 4 shows the UHPLC–QqQ–MS/MS chromatograms for two positive samples containing imidacloprid at 15.22 μg kg−1 (S1) and acetamiprid at 3.56 μg kg−1 (S15).

UHPLC–QqQ–MS/MS extracted chromatograms of two positive real milk samples showing quantification (upper) and confirmation (lower) transitions containing a imidacloprid at 15.22 μg kg−1 (S1) and b acetamiprid at 3.56 μg kg−1 (S15)
Conclusions
In this paper, a simple and sensitive multi-analyte method has been developed for the simultaneous determination of 42 pesticides and six mycotoxins in milk by UHPLC–QqQ–MS/MS, using an SPE-based methodology. Several multi-residue and generic methods were also evaluated, although any of them provided adequate results for AFM1. The widespread QuEChERS method (using acetate and citrate buffers) and the “dilute-and-shoot” procedures evaluated were not suitable for the extraction of AFM1 at very low levels. The application of pre-concentration stages did not improve these results, probably due to suppression signal problems because of the higher matrix amount injected. Therefore, the need for concentration in order to improve the figures of merit for AFM1 led to the application of an SPE methodology that allowed pre-concentration without increasing the amount of matrix in the extract. Furthermore, the use of UHPLC–QqQ–MS/MS improves sensitivity and resolution, detecting and quantifying several classes of compounds satisfactorily. The method showed satisfactory validation parameters, including trueness, precision, inter-day precision, LOD, and LOQ.
The optimized extraction procedure allowed the pre-concentration of the compounds, obtaining LOQ values below the EU tolerance levels. Some advantages of the developed method include simple pre-treatment, rapid determination, and high sensitivity. Furthermore, it could be used in routine analysis for the simultaneous detection and quantification of mycotoxins and pesticides from milk samples.
References
Perseo Program, Spanish Ministry of Health and Consumption/Spanish Food Safety and Nutrition Authority (Ministerio de Sanidad y Consumo/Agencia Española de Seguridad Alimentaria y Nutrición). Available at http://www.perseo.aesan.msps.es/. Accessed on October 2010
Moss MO (2002) Int Biodeterior Biodegrad 50:137–142
MacLachlan DJ, Bhula R (2008) Aust J Exp Agric 48:589–598
Fink-Gremmels J (2008) Food Addit Contam A 25:172–180
Seccia S, Fidente P, Montesano D, Morrica P (2008) J Chromatogr A 1214:115–120
Hussein HS, Brasel JM (2001) Toxicology 167:101–134
Díaz S, Domínguez L, Prieta J, Blanco JL, Moreno MA (1995) J Agric Food Chem 43:2678–2680
Bascarán V, Hernández de Rojas A, Chouciño P, Delgado T (2007) J Chromatogr A 1167:95–101
González-Osnaya L, Soriano JM, Moltó JC, Mañes J (2008) Food Chem 108:272–276
Sørensen LK, Elbæk TH (2005) J Chromatogr B 820:183–196
Commission Regulation (EC) No 839/2008 of 31 July 2008 amending Regulation (EC) No 395/2005 of the European Parliament and of the Council as regards Annexes II, III and IV on maximum residue levels of pesticides in or on certain products. Official Journal of the European Union L234/1, 30 Aug 2008
US Food and Drug Administration (1996) Sec. 527.400 Whole milk, low fat milk, skim milk-aflatoxin M1 (CPG 7106.210). In: FDA compliance policy guides. FDA, Washington, DC, p. 219
Commission Regulation (EC) No 165/2010 of 26 February 2010 amending Regulation (EC) No 1881/2006 setting maximum levels for certain contaminants in foodstuffs as regards aflatoxins. Official Journal of the European Union, L50/8, 27 Feb 2010
Chen CY, Li WJ, Peng KY (2005) J Agric Food Chem 53:8474–8480
Manetta AC, Di Giuseppe L, Giammarco M, Fusaro I, Simonella A, Gramenzi A, Formigoni A (2005) J Chromatogr A 1083:219–222
Boudra H, Barnouin J, Dragacci S, Morgavi DP (2007) J Dairy Sci 90:3197–3201
Decastelli L, Lai J, Gramaglia M, Monaco A, Nachtmann C, Oldano F, Ruffier M, Sezian A, Bandirola C (2007) Food Control 18:1263–1266
Bogialli S, Curini R, Di Corcia A, Laganà A, Stabile A, Sturchio E (2006) J Chromatogr A 1102:1–10
Khay S, Abd El-Aty AM, Choi JH, Shin EH, Kim JS, Chang BJ, Lee CH, Shin SC, Jeong JY, Shim JH (2009) J Sep Sci 32:244–251
Bogialli S, Curini R, Di Corcia A, Laganà A, Nazzari M, Tonci M (2004) J Chromatogr A 1054:351–357
Dagnac T, García-Chao M, Pulleiro P, García-Jares C, Llompart M (2009) J Chromatogr A 1216:3702–3709
Zhu L, Huey Ee K, Zhao L, Kee Lee H (2002) J Chromatogr A 963:335–343
Basheer C, Kee Lee H (2004) J Chromatogr A 1047:189–194
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) J AOAC Int 86:412–431
Cunha SC, Lehotay SJ, Mastovska K, Fernándes JO, Oliveira MBPP (2007) J Sep Sci 30:620–632
Romero-González R, Garrido Frenich A, Martínez Vidal JL (2008) Talanta 76:211–225
Lacina O, Urbanová J, Krplová A, Hajšlová J (2008) Chem Listy 102:s404–s405
Mol HGJ, Plaza-Bolaños P, Zomer P, de Rijk TC, Stolker AAM, Mulder PPJ (2008) Anal Chem 80:9450–9459
Frenich AG, Martínez Vidal JL, Romero-González R, Aguilera-Luiz MM (2009) Food Chem 117:705–712
Beltrán E, Ibáñez M, Sancho JV, Hernández F (2009) Rapid Commun Mass Spectrom 23:1801–1809
Zhang K, Wong JW, Hayward DG, Sheladia P, Krynitsky AJ, Schenck FJ, Webster MG, Ammann JA, Ebeler SE (2009) J Agric Food Chem 57:4019–4029
CEN Standard Method EN 15662: Food of plant origin-determination of pesticide residues using GC-MS and/or LC-MS/MS following acetonitrile extraction/portioning and clean-up by dispersive SPE-QuECHERS method. Available at http://www.cen.eu. Accessed on October 2010
AOAC Official Method 2007.01. Pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulphate
Lehotay SJ, Mastovska K, Yun SJ (2005) J AOAC Int 88:630–638
Commission Decision 2002/657/EEC of 12 August 2002 implementing Council Directive 92/23/EC concerning the performance of analytical methods and the interpretation of the results (2002) Official Journal of the European Communities L221, 17 Aug 2002, pp 8–36
European Commission Directorate General Health and Consumer Protection, Guidance Document on Method Validation and Quality Control Procedures for Pesticide Residues Analyses in Food and Feed, SANCO/10684/2009, 01 Jan 2010
http://ec.europa.eu/sanco_pesticides/public/index.cfm. Accessed on October 2010
Cuadros-Rodríguez L, García-Campaña A, Jiménez-Linares C, Alés-Barrero F, Román-Ceba M (1995) J AOAC Int 78:471–476
British Crop Protection Council (2005–2006) The e-Pesticide Manual. Version 3.2. 13th edn. British Crop Protection Council, Hampshire
Acknowledgements
The authors gratefully acknowledge the Spanish Ministry of Science and Innovation (MICINN-FEDER) for financial support (Project Ref. AGL2006-12127-C02-01 and CTQ2009-07686). MMAL acknowledges her grant (F.P.U.) from the Spanish Ministry of Science and Innovation (Ref. AP2008-02811). PPB is grateful for personal funding through Juan de la Cierva Program (Spanish Ministry of Science and Innovation-European Social Fund, SMSI-ESF). RRG is also grateful for personal funding through the Ramón y Cajal Program (SMSI-ESF).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aguilera-Luiz, M.M., Plaza-Bolaños, P., Romero-González, R. et al. Comparison of the efficiency of different extraction methods for the simultaneous determination of mycotoxins and pesticides in milk samples by ultra high-performance liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 399, 2863–2875 (2011). https://doi.org/10.1007/s00216-011-4670-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-4670-7