
Abstract
An analytical method for multi-class pharmaceuticals determination in wastewater has been developed and validated. Target compounds were: sulfonamides (sulfadiazine, sulfaguanidine, sulfamethazine, sulfamethoxazole), fluoroquinolones (ciprofloxacin, enrofloxacin, norfloxacin), diaminopyrimidine (trimethoprim), anaesthetic (procaine), anthelmintic (praziquantel and febantel), and macrolide (roxithromycin). The method involves pre-concentration and clean-up by solid-phase extraction (SPE) using Strata-X extraction cartridges at pH 4.0. Target analytes were identified and quantitatively determined by liquid chromatography–tandem mass spectrometry using multiple reaction monitoring (MRM). Recoveries were higher than 50% with relative standard deviation (RSD) below 18.3% for three concentrations. Only for sulfaguanidine was low recovery obtained. Matrix effect was evaluated using matrix-matched standards. The method detection limit (MDL) was between 0.5 and 5 ng L−1 in spiked water samples. The precision of the method, calculated as relative standard deviation, ranged from 0.5 to 2.0% and from 1.4 to 8.3 for intra-day and inter-day analysis, respectively. The described analytical method was used for determination of pharmaceuticals in effluent wastewaters from the pharmaceutical industry.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pharmaceutically active compounds (PhACs) are complex molecules with different physicochemical and biological properties and functionality. They have an important role in the treatment and prevention of disease in both humans and animals. Because of their nature they can also have unintended effects on animals and micro-organisms in the environment. Although the effects of pharmaceuticals are investigated in safety and toxicology studies, the potential environmental impacts of their production and use are less understood and have recently become a topic of research interest [1, 2].
The main concern regarding PhACs is that they are being introduced continuously into water bodies as pollutants, and, because of their biological activity, this can lead to adverse effects in aquatic ecosystems and potentially affect drinking water supplies. The main points of collection and subsequent release of pharmaceuticals into the environment are wastewater-treatment plants (WWTPs), where they enter via domestic and hospital sewage or through industrial discharges. Studies of effluent waters and river sediment show that wastewater treatment achieves only partial removal of organic pollutants [3, 4]. Göbel et al. [5] observed high concentrations of several antibiotics (some sulfonamides, macrolides, and trimethoprim) in effluent samples. Some PhACs (tetracyclines, fluoroquinolones, cholesterol-lowering statin drugs, β-blockers) are partially degraded in wastewater-treatment plants, with moderate removal efficiency. Only non-steroidal anti-inflammatory drugs removed with high efficiency (>80%) [6]. As a result, pharmaceuticals are found in surface, ground, and drinking waters [7, 8] and in river sediments and wastewater sludge [9].
Although the concentrations of PhACs are too low to pose an acute risk, it is not known whether other receptors in non-target organisms are sensitive to individual residues, or whether drugs that share a common mechanism of action exhibit synergetic effects [10]. However, because of the widespread usage of PhACs in everyday life and because their purpose is to produce specific biological effects on organisms or living tissue, unwanted environmental effects are to be expected [11]. Because pharmaceuticals are continually being introduced into the environment, they do not need to be persistent to cause negative effects [12]. Nevertheless, there is limited knowledge of the fate and effect of PhACs in the environment and they have not yet been included in any environmental regulation.
Despite the fact that numerous pharmaceuticals have been detected in the aquatic environment, there is still need for new reliable analytical methods. Multiresidue analytical methods are a prerequisite for providing reliable data on the behaviour of pharmaceuticals in the environment and in WWTPs. The information obtained from analysis of influents and effluents of WWTPs may serve to optimize a treatment process or possible pre-treatment step so that emission of undesired pollutants into receiving waters is prevented.
The most common procedures for determination of PhACs in aquatic environmental matrices consist in preconcentration by solid-phase extraction (SPE) followed by separation and determination using liquid (LC) or gas chromatography (GC). The main drawback of GC for PhACs analysis is that this technique is limited to volatile and thermally stabile compounds. Because most pharmaceuticals are polar substances, they need to be derivatized before injection into the GC [13]. Most current analytical methods for separation and detection of pharmaceuticals use liquid chromatography coupled with mass spectrometry (LC–MS). LC with a single-quadrupole MS analyser is highly sensitive, but when very complex matrices such as wastewater are investigated, insufficient selectivity often impairs unequivocal identification of the analytes. Tandem MS affords superior performance in terms of sensitivity and selectivity in comparison with single-quadrupole instruments. Therefore liquid chromatography–tandem mass spectrometry (LC–MS–MS) is the technique of choice for determining polar pharmaceuticals in environmental samples [14, 15].
Several analytical methods are available in the literature for the determination of pharmaceutical compounds in surface and wastewaters, and numerous papers have reported the occurrence of specific pharmaceuticals at levels ranging from ng L−1 to μg L−1 but the vast majority of these are focused on specific therapeutic classes, paying special attention to antibiotics which are believed to be the ones of biggest concern. Multi-residue analytical methods, including multiple-class pharmaceuticals, are becoming the required tools to provide reliable and wider knowledge about their occurrence, and for the monitoring of their removal. Simultaneous analysis of several groups of compounds with quite different physicochemical characteristics generally requires a compromise in the selection of experimental conditions, which in some cases means not obtaining the best performance for each of the compounds. However, developing a multi-group method is rewarding as it can be applied in routine analysis, providing a large amount of data.
The objective of this work was to develop a sensitive multi-class analytical method for determination of 12 pharmaceuticals in wastewater from the pharmaceutical industry. Target compounds were selected on the basis of pharmaceutical industry production and belong to different structural groups. Chemical structures of the PhACs investigated are shown in Fig. 1.

Chemical structures of the pharmaceuticals studied
The developed method involves sample pre-treatment by solid-phase extraction (SPE) followed by high-performance liquid chromatography (HPLC) separation and tandem mass spectrometric (MS–MS) detection carried out using an electrospray (ESI) interface. From the observed ion fragmentation pathways a reliable and sensitive quantification method was developed. The performance of the method was evaluated by estimation of the linearity, sensitivity, repeatability, reproducibility, and matrix effects. Finally, the method was successfully applied to analysis of PhACs in pharmaceutical industry wastewaters.
Experimental
Pharmaceutical standards and reagents
The pharmaceuticals studied were: praziquantel (PRAZ), febantel (FEBA), trimethoprim (TMP), norfloxacin (NOR), ciprofloxacin (CIPRO), enrofloxacin (ENRO), sulfaguanidine (SGUA), sulfadiazine (SDIAZ), sulfamethazine (SMETH), sulfamethoxazole (SMETOX), procaine (PROC), and roxithromycin (ROXI). High-purity (>99%) analytical standards of the drugs were obtained from Veterina Animal Health (Kalinovica, Croatia) (PRAZ, FEBA, TMP, ENRO, SGUA, SDIAZ, SMETH, and PROC) or supplied by Sigma–Aldrich (NOR, CIPRO, SMETOX, and ROXI). The pharmaceuticals studied are shown in Fig. 1 and their physicochemical properties are listed in Table 1.
A stock solution of the pharmaceutical mixture was prepared by dissolving accurately weighed quantities of the powdered standards in acetonitrile–water 1:1 (v/v) and stored away from light at 4 °C. The mass concentrations of each pharmaceutical in the stock solution were: 10 μg mL−1 for PRAZ and FEBA; 50 μg mL−1 for ENRO, SMETOX, SMETH, and TMP; and 100 μg mL−1 for SGUA, SDIAZ, NOR, CIPRO, PROC, and ROXI. Working standard solutions were prepared from this stock by serial dilution. For the pH-value adjustment of water samples, hydrochloric acid solution (0.1 mol L−1) was used. All the solvents used were HPLC-grade supplied by Kemika (Zagreb, Croatia).
Sample pre-treatment and SPE
The method was optimized and validated using pharmaceuticals-free water. The pharmaceuticals-free water sample was collected from the wellwater Borčec, near Zagreb, Croatia. Wellwater samples were pre-filtered through 0.45 μm filters to eliminate particulate matter.
Wastewater samples were collected from wastewater-treatment plants of the pharmaceutical industry. Once in the laboratory, wastewater samples were filtered through a Büchner funnel (black Whatman) followed by 0.45 μm Nylon membrane filters.
Amber glass bottles pre-rinsed with ultra-pure water were used for sample collection. All water samples were stored at 4 °C until SPE extraction, which was performed within 24 h.
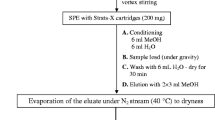
The pharmaceuticals studied were extracted from water samples and pre-concentrated on 500 mg/3 mL Strata-X cartridges supplied by Phenomenex (Torrance, CA, USA) using an apparatus for solid-phase extraction. Before water application, cartridges were conditioned with 5 mL each of methanol, and water. The pH of samples and water for preconditioning was adjusted to 4.0 with hydrochloric acid solution (0.1 mol L−1). A sample volume of 100 mL was applied to the cartridge and the flow was kept at no greater than 4 mL min−1. The cartridge was left to dry for approximately 5 min, using vacuum to remove excess water. Analytes retained were eluted with 2 × 5 mL methanol. Following elution, filtrates were evaporated to dryness on a rotary evaporator at 40 °C and redissolved in 1 mL acetonitrile–water 1:1 (v/v), achieving 100-fold preconcentration.
For determination of recovery during SPE, pre-filtered pH adjusted wellwater was spiked with appropriate amounts of the analytes.
LC–ESI-tandem MS analysis
LC analysis was performed using an Agilent (Santa Clara, CA, USA) Series 1200 HPLC system equipped with a Synergy Fusion C18 embedded column (150 mm × 2.0 mm, particle size 4 μm) supplied by Phenomenex. The analysis was performed using 0.1% formic acid in MilliQ water as eluent A and 0.1% formic acid in acetonitrile as eluent B in gradient elution mode; the gradient conditions are listed in Table 2. After gradient elution, the column was equilibrated for 12 min before another injection. Flow rate was 0.2 mL min−1. An injection volume of 5 μL was used in all analysis.
Tandem MS analysis was carried out on an Agilent 6410 triple-quadrupole mass spectrometer equipped with an ESI interface. The analyses were done in positive-ion (PI) mode for all the analytes investigated. The conditions for the analyses were: drying gas temperature 350 °C; capillary voltage 4.0 kV; drying gas flow 11 L min−1 and nebulizer pressure 35 psi. Following selection of the precursor ions, product ions were obtained at a series of collision energies and selected according to the fragmentation that produced a useful abundance of fragment ions. The optimum collision energies, fragmentor voltages, and transitions chosen for the multiple reaction monitoring (MRM) experiment are listed in Table 3.
Instrument control, data acquisition and evaluation were done with Agilent MassHunter 2003–2007 Data Acquisition for Triple Quad B.01.04 (B84) software.
Validation of analytical procedure
Each compound was analysed by MRM, using the two highest characteristic precursor ion/product ion transitions. Positive identification criteria for the target analytes were based on LC retention time and the ratio of the abundances of two specific precursor ion/product ion transitions.
The extraction recoveries of the target compounds were determined using wellwater samples spiked with the analytes at three concentrations. Recoveries were determined by comparing the concentrations obtained with the initial spiking levels. In each case, samples were analysed in triplicate.
The linearity of the method was evaluated by using wellwater spiked with the target pharmaceuticals. The precision of the method was determined by repeated (n = 3) intra-day and inter-day analysis of spiked wellwater at a concentration in the middle of the linear range. The precision of the method was expressed as the relative standard deviation (RSD) of replicate measurements. Method detection limits (MDL) and method quantification limits (MQL) were determined, by analysis of spiked wellwater samples, as the concentrations that would give S/N ratios of 3 and 10, respectively.
When experiments with wastewaters were performed, matrix-matched standards were prepared by spiking wastewater samples. Because unspiked WWTP effluent already contained some of the investigated compounds, analyte concentration was calculated by subtracting the concentration for these analytes in the matrix from the spiked concentration.
Results and discussion
SPE procedure
The critical step in method development is the sample-preparation procedure. The choice of sorbent is a key point in solid-phase extraction (SPE) because it can affect method performance, for example selectivity, affinity, and capacity. The choice depends strongly on the analytes of interest and the interactions of the chosen sorbent with the functional groups of the analytes. However, it also depends on the kind of a sample matrix and its interactions with both the sorbent and the analytes [18]. For water samples with analytes of different polarity, polar analytes complicate the choice of the most appropriate SPE sorbent [19]. In addition to the polarity range of the analytes, the pH of water samples has a significant effect, especially for amphoteric compounds [20]. As can be seen from Fig. 1, all the sulfonamides investigated have at least two nitrogen functions. The amide attached to the sulfur is referred to as N1 and is deprotonated at pH 6.5–7.4 (except for sulfaguanidine, 11.3). The amine attached to the aromatic ring is referred to as N4 and is protonated at pH 2.5. For this reason, sulfonamides are positively charged under acidic conditions, are neutral between pH 2.5 and 6, and are negatively charged under alkaline conditions. Trimethoprim is, also, characterized by two pK a values corresponding to protonation of the two heterocyclic nitrogen atoms (N1 and N3). Fluoroquinolones are cationic under acidic conditions because of deprotonation of the carboxylic acid group and protonation of the piperazinyl amino group (N4). Because sulfonamides are also amphoteric, the pH of the sample solution is one of the most important conditions to control.
The pharmaceuticals selected (Fig. 1) have different polarity (log K ow shown in Table 1) and most of them have amphoteric character (pK a values shown in Table 1), especially sulfonamide and fluoroquinolone pharmaceuticals. Because of these considerations it is very important to select right water sample pH and appropriate SPE sorbent material. To optimise the extraction procedure the pH of SPE-loaded water samples was investigated. Based on preliminary experiments and recovery experiments at different pH, pH 4.0 was selected [21]. The Strata-X SPE cartridge selected has a surface-modified styrene skeleton with a pyrrolidone group, whose retention mechanisms are hydrophobic, hydrogen-bonding, and aromatic. This sorbent is used for reversed-phase extraction of acidic, basic, and neutral compounds. In several papers, Strata-X sorbent has been compared with other commercially available sorbents for retention of polar and non-polar compounds from water samples [22, 23], and was found to be the best solid phase for extraction of number of analytes by off-line SPE.
Recovery was studied by spiking wellwater with a mixture of pharmaceuticals. Experiments were performed to address the SPE cartridge breakthrough properties of each of individual compound by using systems with pharmaceuticals in mixtures at three concentrations. Recoveries of pharmaceuticals were expressed as average values from three determinations. Mean recoveries obtained at each concentration are detailed in Table 4.
Ideally, the extraction recovery should not be sample-concentration-dependent. In other words, for the method to be useful there should be no significant difference in recovery over the expected concentration ranges of the compound to be analysed. Recovery results for the analysed pharmaceuticals in mixtures show that total pharmaceutical concentrations affect cartridge capacity for individual compounds. The data in Table 4. indicate that in experiments when the overall concentrations of compounds increase, a compound that is adsorbed relatively weakly by the solid phase may be displaced by a more strongly adsorbed component, which may result in a breakthrough concentration for displaced compounds that exceeds the concentration in the mixture.
Recoveries (Table 4) obtained for all target pharmaceuticals, using 500 mg/3 mL Strata-X cartridges were higher than 50% (at all concentrations), except for sulfaguanidine (25.6–38.2%). As already mentioned, the mixture investigated contains pharmaceuticals of different polarity (log K ow between −1.22 and 2.75) which compete for places on the cartridges. In this competition, less polar compounds are adsorbed most strongly by the cartridge. The most hydrophobic of the pharmaceuticals investigated is roxithromycin (Table 1), the largest compound, and with a branched structure (Fig. 1). Because of this, it is strongly retained and its functional groups and branched structure could obstruct the retention of other pharmaceuticals. (e.g. CIPRO, NOR, FEBA, and TMP). The negative effect of ROXI on the extraction efficiency, in particular for the less polar compounds, can be attributed also to interaction of other analytes with this substance, and not only because of coverage of the binding sites by adsorption of ROXI on the sorbent. H-bonding plays a vital role in adsorption of organic compounds, and covalent bonding involving carbonyl, and carboxyl groups has been also reported. Aromatic amines in aqueous solutions can be bound to dissolved organic substances, for example ROXI, by reversible and irreversible reaction mechanisms, which can partly explain their low recovery.
Sulfaguanidine as the smallest molecule and the most hydrophilic compound is least strongly adsorbed by Strata X and its recovery is not affected by increasing the total concentration of pharmaceuticals. Furthermore, the appropriate sorbent mass for a given extraction provides sufficient capacity to retain both the analyte and any contaminants that may also be retained during the loading step. Insufficient sorbent mass leads to column overload and low or irreproducible recoveries. Increasing the analyte concentrations also affects the capacity of the sorbent and requires more sorbent mass. But it is important to say that more sorbent mass demands increased solvent requirements and may also reduce recovery.
LC-ESI-tandem MS analysis
Chromatographic separation of the selected pharmaceuticals was achieved on an embedded C18-modified chromatographic column. Improvement of the separation between all the mixture components was achieved by changing the solvents in mobile phase (formic acid in acetonitrile–methanol as organic phase and formic acid in water as inorganic phase were used), the volume proportion of the solvents in mobile phase, and the concentration of formic acid (0.01% and 0.1%). Further improvement in separation were obtained by use of a mobile phase gradient and by changing the flow rate.
For all the compounds analysed better ionization was achieved with 0.1% formic acid and use of electrospray ionization in positive-ion mode. In subsequent experiments methanol was rejected as an option for the organic phase because of broadening of chromatographic peaks. When the flow rate was changed, grouping of all the pharmaceuticals at the start of the chromatogram was observed at higher flow rates (0.5 mL min−1 and 0.7 mL min−1). Better separation was achieved at lower mobile phase flow rates (0.1 mL min−1, 0.2 mL min−1, and 0.3 mL min−1). So, the best separation of all the investigated pharmaceuticals was obtained with 0.2 mL min−1. The mobile phase gradient was started with 0% organic phase because sulfaguanidine is the smallest and the most polar pharmaceutical in the investigated mixture. Otherwise, all gradients with even the lowest content of organic phase caused overlapping of the sulfaguanidine peak with that of the mobile phase. Application of gradient elution as described in the experimental section gave a good chromatographic separation of the analytes with R t values in the range 2.7 to 23.5 min. A representative chromatogram obtained from spiked wellwater is shown in Fig. 2.

Chromatograms obtained from spiked wellwater: total ion chromatogram (a) and extracted and overlapped MRM chromatograms for the target analytes (b)
The ESI interface conditions were optimized for all individual components in the PI mode in order to obtain the best instrumental conditions for identification of the target pharmaceuticals. The protonated molecules, [M+H]+, were the base peaks for all the compounds and were selected as precursor ions. After selection of precursor ions, MS-MS spectra in product ion mode of operation were acquired to obtain information about fragment ions. When the product ions had been selected for every analyte, MRM experiments were carried out to select the optimum collision energy for every specific transition. Collision energies in the range 10 to 35 eV were investigated. The optimum energies were those that gave the best sensitivity for every transition. Table 3 summarizes the most relevant MS settings, for example fragmentor voltages and collision energies, used for each pharmaceutical.
The most intense fragment ion from each precursor ion was selected as transition ion for detection and quantification. For this purpose, two criteria for positive identification were used:
-
1.
correlation of retention times with those of standards (±2%); and
-
2.
the first selected precursor/product ion transition.
The second most intense transition was used for confirmation purposes.
Matrix effect
One significant drawback of ESI-MS is that the ionisation source is highly susceptible to other compounds present in the matrix. The matrix effect typically results in suppression or enhancement of the analyte signal. Therefore, LC–MS–MS signal response obtained from standard and matrix samples may differ significantly [24, 25].
The main source of ion suppression is the presence of endogenous substances, i.e. organic or inorganic molecules present in the sample and that are retrieved in final extract. When analysing wastewater samples a source of ion suppression is natural dissolved organic matter, which is a complex mixture of naturally occurring organic compounds. A second source is the presence of exogenous substances, i.e. molecules not present in the sample but coming from various external sources during the sample-preparation procedure (for example, material released by SPE) [24].
The consequences of ion suppression are numerous: detection capability is reduced; repeatability is affected because of variable degrees of ion suppression for different samples, and linearity, ion ratio, and quantification are affected. Therefore, ion suppression may lead to failure to detect an analyte which is present, or underestimation of its real concentration, or to non-fulfilment of the identification criteria with consequences in terms of false-negative results. On the other hand, ion enhancement may lead to overestimation of the analyte concentration with a risk of false positive result [24].
Matrix effects were evaluated to assess their effect on quantification of the pharmaceuticals in this investigation, where the sample is a complex matrix. In order to evaluate the degree of ion suppression or enhancement, the ratio of the analyte signal from wastewater to the analyte signal from solvent (acetonitrile–water 1:1 (v/v)) was studied. Blank samples were evaluated simultaneously in order to subtract concentrations of analytes present in the sample. For all compounds except praziquantel and procaine some degree (<30%) of signal suppression (CIPRO, NOR, ENRO, FEEBA, and TMP) or enhancement (ROXI, SGUA, SDIAZ, SMETOX, and SMETH) was observed.
Method validation
The performance characteristics of the SPE–LC–MS–MS method were established by validation with spiked water samples. Linearity, method limits of detection (MDL) and quantification (MQL), precision, and recovery were evaluated for quantitative purposes. The linearity was evaluated for each pharmaceutical in the mixture using five concentrations in the range of 5–10,000 ng L−1 depending on the pharmaceutical. Calibration curves were prepared for each compound from the spiked wellwater by plotting peak area versus analyte concentration. Blanks were also prepared as a quality control tool, but not included in regression analysis. The results were analysed by linear regression. Correlation coefficients were higher than 0.9926 except for trimethoprim (0.9870) thus confirming the linearity of the method (Table 5).
Method limits of detection (MDL) and quantification (MQL) were experimentally estimated for each pharmaceutical using signal-to-noise ratios from mass chromatograms obtained in SRM mode for spiked wellwater samples. Detection and quantification limits obtained are reported in Table 5. MDL ranged from 0.5 to 5 ng L−1 and MQL were from 5 to 50 ng L−1. To ensure correct quantification, the precision of the method was studied by analysing three replicates of the standard with a concentration in the middle of the linear range. The results obtained are listed in Table 5, and show that intra-day precision was from 0.5 to 2.0% and inter-day precision was from 1.6 to 8.3%.
Application of the method
The described method was applied to determination of the target pharmaceuticals in two wastewater samples from the pharmaceutical industry. Wastewater treatment consisted of only primary settlement and the plant receives only industrial wastewater. The chemical synthesis processes of the veterinary pharmaceuticals industry produce wastewaters which are variable in character depending on production. Three wastewater samples (wws), collected during two months, were analysed. The results are summarized in Table 6.
Enrofloxacin, praziquantel, sulfaguanidine, sulfadiazine, and sulfamethazine were detected in all the wastewater samples, as shown in the chromatograms obtained (Fig. 3). Norfloxacin, roxithromycin, procaine, sulfamethoxazole, and trimethoprim were not detected. Praziquantel and febantel were detected at the lowest concentrations in wastewater. Sulfadiazine was detected in wastewater samples 2 and 3 at very high concentration.

Chromatograms obtained from wastewater sample 2 (wws2)
Conclusion
This multi-class method, based on SPE followed by LC–MS–MS determination is proposed for simultaneous analysis of 12 pharmaceutical compounds with a variety of structures and different physicochemical properties: sulfadiazine, sulfaguanidine, sulfamethazine, sulfamethoxazole, ciprofloxacin, enrofloxacin, norfloxacin, trimethoprim, procaine, praziquantel, roxithromycin, and febantel. Recoveries obtained for investigated pharmaceuticals, using Strata-X cartridges, were higher than 50% (at three concentrations), except for sulfaguanidine. The compounds were analysed by LC–MS–MS in positive-ionisation mode, operating in MRM mode, with two transitions monitored for each analyte; this provided good sensitivity and selectivity, with detection limits in the low ng L−1 range in spiked water samples.
The method was successfully applied to the analysis of wastewater samples from the pharmaceutical industry so it can be useful tool for determination of the amounts of these pharmaceuticals discharged from WWTPs into the aquatic environment and for evaluation of the effect of conventional WWTPs in the elimination of pharmaceutical compounds.
References
Kümmerer K (2008) Pharmaceuticals in the environment. Springer, Berlin
Daughton CG, Ternes T (1999) Environ Health Perspect 107:907–938
Ternes TA (2001) Trends Anal Chem 20:419–434
Carballa M, Omil F, Lema JM, Llompart M, García-Jares C, Rodríguez I, Gómez M, Ternes T (2004) Water Res 38:2918–2926
Göbel A, McArdell CS, Joss A, Siegrist H, Giger W (2007) Sci Total Environ 372:361–371
Gros M, Petrović M, Ginebreda A, Barceló D (2010) Environ Int 36:15–26
Gros M, Petrović M, Barceló D (2006) Talanta 70:678–690
Grujić S, Vasiljević T, Laušević M (2009) J Chromatogr A 1216:4989–5000
Jelić A, Petrović M, Barceló D (2009) Talanta 80:363–371
Halling-Sørensen B, Nielsen SN, Lanzky PF, Ingerslev F, Lützhøft HCH, Jørgensen SE (1998) Chemosphere 36:357–394
Hernando MD, Petrović M, Fernández-Alba AR, Barceló D (2004) J Chromatogr A 1046:133–140
Radjenović J, Petrović M, Barceló D (2007) Trends Anal Chem 26:1132–1144
Reeves VB (1999) J Chromatogr B 723:127–137
Petrović M, Hernando MD, Díaz-Cruz MS, Barceló D (2005) J Chromatogr A 1067:1–14
Kot-Wasik A, Dębska J, Namieśnik J (2007) Trends Anal Chem 26:557–568
EPIweb 4.0 (http://www.epa.gov/oppt/exposure/pubs/episuitedl.htm), February 2010
Babić S, Horvat AJM, Mutavdžić Pavlović D, Kaštelan-Macan M (2007) Trends Anal Chem 26:1043–1061
Mutavdžić Pavlović D, Babić S, Horvat AJM, Kaštelan-Macan M (2007) Trends Anal Chem 26:1062–1075
Weigel S, Kallenborn R, Huhnerfuss H (2004) J Chromatogr A 1023:183–195
Renew JE, Huang CH (2004) J Chromatogr A 1042:113–121
Mutavdžić D, Babić S, Ašperger D, Horvat AJM, Kaštelan-Macan M (2006) JPC-J Planar Chromat 19:454–462
Mutavdžić Pavlović D, Babić S, Dolar D, Ašperger D, Košutić K, Horvat AJM, Kaštelan-Macan M (2010) J Sep Sci 33:258–267
Hilton MJ, Thomas KV (2003) J Chromatogr A 1015:129–141
Antignac J-P, de Wasch K, Monteau F, De Brabander H, Andre F, Le Bizec B (2005) Anal Chim Acta 529:129–136
José Gómez M, Petrović M, Fernández-Alba A, Barceló D (2006) J Chromatogr 1114:224–233
Acknowledgements
This work was supported by the Unity Through Knowledge Fund (UKF), which was established by the Croatian Ministry of Science, Education and Sports through World Bank Loan No. 7320-HR: Reduction of environmental risks posed by pharmaceuticals and their degradation products in process wastewaters, through RO/NF membrane treatment (REPHAD), and Croatian Ministry of Science, Education and Sports Projects: 125-1253008-1350 and 125-2120898-3148.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Babić, S., Mutavdžić Pavlović, D., Ašperger, D. et al. Determination of multi-class pharmaceuticals in wastewater by liquid chromatography–tandem mass spectrometry (LC–MS–MS). Anal Bioanal Chem 398, 1185–1194 (2010). https://doi.org/10.1007/s00216-010-4004-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-4004-1