
Abstracts
Self-assembled monolayers (SAMs) provide a convenient, flexible and simple system to tailor the interfacial properties of metals, metal oxides and semiconductors. Monomolecular films prepared by self-assembly are attractive for several exciting applications because of the unique possibility of making the selection of different types of terminal functional groups and as emerging tools for nanoscale observation of biological interactions. The tenability of SAMs as platforms for preparing biosurfaces is reviewed and critically discussed. The different immobilization approaches used for anchoring proteins to SAMs are considered as well as the nature of SAMs; particular emphasis is placed on the chemical specificity of protein attachment in view of preserving protein native structure necessary for its functionality. Regarding this aspect, particular attention is devoted to the relation between the immobilization process and the electrochemical response (i.e. electron transfer) of redox proteins, a field where SAMs have attracted remarkable attention as model systems for the design of electronic devices. Strategies for creating protein patterns on SAMs are also outlined, with an outlook on promising and challenging future directions for protein biochip research and applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The design and structural organization of biointerfaces suitable for molecular recognition and mapping of protein interaction networks are keys for obtaining protein arrays with high performance characteristics [1–3]. In addition, many experimental approaches in biological studies, clinical diagnostic, drug discovery and proteomics require the immobilization of proteins on surfaces [4–9]. Proteins can also act as recognition elements in sensor devices where a protein is attached to a solid support and the combined unit acts as an optical or electronic transducer. In particular, the success achieved by DNA arrays in genomics stimulated scientific interest in the use of protein arrays as tools in proteomics [5, 8]. However, since proteins are much more complicated from both a chemical and a structural point of view than nucleic acids, their immobilization on surfaces is intrinsically more complicated [10–13]. Additional difficulties derive from the sensitive nature of proteins, which have low conformational stability and can be damaged upon surface immobilization [11, 12]. Moreover, proteins can adhere and adsorb to most surfaces through a variety of mechanisms (electrostatic interaction, hydrogen bonding, hydrophobic interaction and a combination of these), resulting in non-specific protein binding [14]. Covalent binding of the protein to a reactive surface through surface-accessible residues often lacks regiospecificity and hence immobilized protein may not be correctly oriented. Additionally, the reactive site of a protein can be blocked by the immobilization procedure, resulting in the reduction or loss of all of the protein activity. Therefore, one of the paramount challenges for a primary enabling technology is to achieve controlled immobilization of the proteins on surfaces in such a way that they retain their biological features and properties and avoiding non-specific protein binding. Molecular surface science has greatly contributed to the improvement of this technology by providing ideal platforms for creating surfaces bioengineered at a molecular level [15]. Among the surface-modification techniques, self-assembly is one of the most popular bottom-up approaches to add the anchoring elements: it is easy to apply and allows one to tune the functionality of the layer created by modifying the end groups.
Self-assembly, involving spontaneous arrangements of atoms and molecules in an ordered functional entity, is one of the main strategies used in nature allowing life to be created from its building blocks [16, 17]. Recently, much effort has been devoted to a fundamental understanding of molecular film self-organized on surfaces [18]. Among various self-assembly processes, the formation of a self-assembled monolayer (SAM) is one of the most elegant ways for making an organic film with specific surface properties [19–21]. Although the Langmuir–Blodgett method allows the transfer of a monolayer from an air–water interface onto a solid support, so that multilayers can be formed simply by repeating the transfer steps, the multilayers are thermodynamically unstable and, consequently, minimal temperature changes or exposure to solvent can ruin their two-dimensional structure [16, 22]. On the other hand, SAM formation does not only give more rugged organic films, as consequence of strong chemisorption of suitable molecules such as organosulfur compounds, but is also capable of imprinting a desired function when individual molecules are assembled into highly oriented and ordered architecture. The aim of this review is to highlight some smart SAM architectures and their novel applications. The design and use of SAMs for application in biomolecular recognition, including the use of monolayers for assembling functional tethered bilayer lipid membranes (t-BLMs), the development of methods to modify the exposed surfaces of SAMs formed from thiols and their dynamic interaction with biomolecules are reviewed. Strategies for creating protein patterns on SAMs and moieties bearing binding sites for specific attachment of target protein are also outlined, with an outlook on promising and challenging future directions for protein biochip research and applications.
Organization and nature of the metal–SAM interface
SAMs are organic assemblies formed when molecules in solution or the gas phase adsorb and spontaneously organize into a single layer on a surface. The molecules constituting SAMs have chemical functionality with a specific affinity for a substrate. There are a number of chemical functionalities for binding to specific metals [21, 23–25], metal oxides [26], semiconductors [27, 28] and glass [29, 30]. The most widely applied class of SAMs derives from the adsorption of an alkanethiol on gold; in fact, it forms good (well-packed) SAMs and it is historically the most studied. Moreover, gold is a reasonably inert metal, easy to prepare and pattern by a combination of lithographic tools and chemical etchants. Gold binds thiols with a high affinity and it does not undergo any unusual reaction with them. In particular, Au(111) yields SAMs having both the highest density and the highest degree of regularity [31]. Nonetheless, this thiol–gold bond leads to facile exchange of the adsorbates. SAMs of alkanethiols on gold provide suitable systems to explore the effect of surface chemistry on protein adsorption and cell adhesion, because thin gold films are common substrates for several analytical techniques, including surface plasmon resonance (SPR) spectroscopy, quartz crystal microbalances (QCMs), ellipsometry and electrochemical methods. In particular, cells can adhere and properly work on gold surfaces without evidence of toxicity [32–34].
SAMs of organosulfur compounds adsorb spontaneously on substrates from either the liquid or the vapour phase. Assembly from solution on the laboratory bench is convenient and sufficient for most applications of SAMs, especially for those requiring contact with other liquid phases in subsequent experiments. In the adsorption of alkanethiols from solution, the desired clean substrate has to be dipped in the required dilute solution of the thiol for a specified time and temperature. Gas-phase evaporation of the adsorbent can also form good monolayers, although structural control can sometimes be difficult. The experimental conditions more frequently adopted for preparing SAMs yield organic interfaces having reproducible and desired functional behaviours. Nevertheless, there are a number of experimental factors that can affect the kinetics of formation and the structure of the resulting SAM, such as the nature and roughness of the substrate, solvent, temperature, concentration of adsorbate, immersion time, oxygen concentration of the solution and nature of the adsorbate. Comprehensive reviews of the practical protocols for preparing SAMs and details of the thermodynamics and kinetics governing the process of assembly are available [19, 20, 35]. It was also reported how easily SAM removal from gold surface can be achieved by cathodic polarization; an alkaline medium constituted by KOH in water or ethanol is usually employed. Both the thiolate and the bare metal surface become solvated, and the thiolate diffuses away from the surface. The process is reversible: removing the applied negative potential can result in readsorption of the thiolates onto the metal surface. A method for patterning multiple types of adherent cells on the same substrate by electrochemical desorption of SAMs in localized areas defined by a microfluidic system has been reported [36]. This technique has the capability to pattern different types of cells with precisely controlled distance, allowing the free exchange of soluble molecules. The manipulation and microfabrication required in this approach can be extensively applied for studying fundamental biomedical problems based on cell–cell interactions. Furthermore, spatially selective electrochemical transformations can find uses in microfabrication and surface engineering [37, 38].
Over the past two decades, comprehensive understanding of the assembly of SAMs evolved considerably as a consequence of the availability of improved or new methods for their characterization. The combination of data from scanning probe microscopes (atomic force microscope, scanning tunnelling microscope, etc.), spectroscopic techniques such as reflection absorption infrared spectroscopy, X-ray photoelectron spectroscopy, and ellipsometry, and physical methods provided a reasonable understanding of the structural organization of SAMs and the assembly process at the molecular level [39–41]. Although many controversies still exist about the dynamics of the assembly process and the structural characteristics of the alkanethiol SAM–gold surface interface, such as the head-group bonding structure, the packing configuration and the nature of the adsorption site [42, 43], it is clear that the process leading to the formation of SAMs involves a subtle interplay of the energetics of the metal–sulfur bonds and noncovalent lateral interactions among the organic groups. Recently, the existence of ion and water channels in highly dense and well-ordered alkanethiol SAMs has received considerable interest [44–46]. In most cases, the specific ordering of the sulfur moieties on the metal lattice defines the free space available to the organic components. The maximization of the attractive lateral interactions (van der Waals, hydrogen bonding) determines the organization of the organic layer. The organic groups can also restrict the density of coverage; steric crowding of the organic groups can limit the arrangement of the sulfur atoms to one layer less dense than that exhibited by elemental sulfur on a given substrate. The formation of alkanethiolates on Au(111) requires the balance between the adlayer structure and the lateral interactions stabilizing the assembly: the metal–sulfur interaction drives the assembly to the limiting case where the gold surface is covered by an overlayer of thiolates. On the other hand, the attractive lateral interactions also promote the secondary organization of the alkane chains, giving a superlattice structure (Fig. 1). To achieve a dense monolayer, several surface-active organosulfur compounds have been studied [20, 21]. These include alkanethiols [HS(CH2) n X], dialkyl disulfides [X(CH2) m S − S(CH2) n X] and dialkyl sulfides [X(CH2) m S(CH2)nX], where n and m are the number of methylene units and X represents the end group of the alkyl chain (−CH3, −OH, −COOH, −NH2, etc.). The evidence available suggests that on gold either thiols or analogous disulfide forms SAMs of similar structure [47, 48]. The low solubility of disulfides makes them difficult to use in solution; this leads to their precipitation and the possible formation of a multilayer contamination of the substrate if the sample preparation does not occur under carefully controlled conditions [23]. Dialkyl sulfides form SAMs that are similar to those formed by thiols and disulfides, but the interaction of sulfur with the metal surface is weaker than the metal thiolate bond, and, thus, the SAMs formed from sulfides are less stable than those formed from thiols and disulfides [49]. As demonstrated by electrochemical [50], X-ray photoelectron spectroscopy [51] and mass-spectrometric [52] characterization of dialkyl sulfide SAMs, any cleavage of the C − S bond is observed during formation; furthermore, scanning tunnelling microscopy studies suggest that the adsorbates are not as well ordered as those derived from thiols or disulfides [53, 54].

The arrangement of decanethiolates on an Au(111) lattice when maximum coverage of the thiolates is attained. a Structural model of the commensurate adlayer formed by thiols on the gold lattice. The arrangement shows a structure where the sulfur atoms (dark-grey circles) are positioned in the threefold hollows of the gold lattice (white circles, a = 2.88 Å). The light-grey circles with the dashed lines indicate the approximate projected surface area occupied by each alkane chain; the dark wedges indicate the projection of the CCC plane of the alkane chain onto the surface. The alkane chains tilt in the direction of their next-nearest neighbours. b Cross-section of the self-assembled monolayer (SAM) formed from decanethiol. Note the alternating rotations of the carbon chains in this view. The chains are labelled with twist values (β) to indicate the relative orientations of the neighbouring chains. (From [21])
The chain length is also important for controlling the density of the monolayer: longer-chain alkanethiols bearing a terminal group [i.e., HS(CH2) n X, where n ≥ 10] assemble into densely packed and ordered monolayers [55]. For example, phenyl and biphenyl SAMs show good packing owing to π–π interactions, but are less stable than those of long-chain alkanethiols [56]. However, although long-chain SAMs are very stable and effective in reducing non-specific interactions with biomolecules [57], they have limited applications in electrochemical sensors, a field where SAMs have attracted remarkable attention as model systems for design of electronic devices [58, 59]. This disadvantage could be overcome using mixtures of alkanethiols of different length [60, 61]. Mixed SAMs are monolayers comprising a well-defined mixture of molecular structures; they provide a method useful for incorporating molecular species whose own physical dimensions would prevent a direct, well-organized assembly. Two specific examples include the formation of SAMs that contain ligands or proteins retaining their active/native conformations [62], and the placement of electroactive species, correctly oriented, at precise distances from the electrode surface [63, 64] (see also the next section). Mixed SAMs are also useful for defining gradients of interfacial composition, which are useful for studying the properties and biology of cells [32]; however, the low immobilization capacity of these surfaces negatively affects the sensor performance [65]. Recently, SAMs of dithiolated scaffolds have been studied as supports for the immobilization of biomolecules [66, 67]. These scaffolds contain two identical alkyl thiol substituents attached to a phenyl ring through phenolate bridges, providing two attachment points on the gold surface; this generates a SAM more stable than the monothiol. Furthermore, it provides a more adequate spacing of an immobilized biomolecule, thus allowing improved mobility and flexibility at the recognition terminus [68]. Such a structure has lower insulating ability than alkanethiol SAMs and therefore is expected to allow easier electron transfer; thus, this kind of dithiol-modified SAM can be profitably used as a support in electrochemical sensors [69].
Randomly and uniformly oriented protein grafting on SAMs: versatile platforms for bioanalytical applications
SAMs of alkanethiols on gold provide suitable systems for studying biological and biochemical processes. The stability, the uniform surface structure and the relative ease of varying their functionalities make SAMs an appealing choice for immobilizing sensing molecules on a transducer surface. In the following, the different methods usually employed for protein immobilization are reported and discussed according to the kind of interaction exploited.
Non-covalent interactions
Conventional grafting of unmodified proteins onto a reactive surface leads to statistical orientation of the proteins on the underlying surface. Non-covalent protein immobilization methods are widely employed and involve either passive adsorption onto hydrophobic surfaces or electrostatic interactions with charged surfaces.
Hydrophobic interactions
Immobilization via hydrophobic interactions on alkanethiol SAMs has been shown to be suitable for certain proteins, such as the blue copper protein azurin [70, 71]. The molecular orientation with the copper redox centre facing the electrode surface has been achieved by using a methyl-terminated alkanethiol SAM on which azurin molecules are immobilized through hydrophobic interactions between the azurin hydrophobic area around the copper centre and the akanethiol methyl heads (Fig. 2). A QCM coupled with electrochemistry was recently used to examine the adsorption of azurin on a gold electrode modified with a SAM of octanethiol [72]. The QCM data revealed that the kinetics of azurin adsorption onto such electrodes could be controlled by the azurin concentration. With only modest azurin concentrations (approximately micromolar) the saturation coverage was attained quite rapidly (less than 3 min): this has implications for experiments where an adsorbed layer of protein is required. The high stability and sensitivity of this system could hold promise for electronic mapping of a single redox protein molecule and protein-based biosensors.

Molecular orientation for azurin with the copper redox centre facing the Au(111) surface, achieved by the hydrophobic adsorption of azurin on methyl-terminated SAMs of alkanethiol. The drawing is not to scale, and the dimensions of some atoms (e.g. Cu, S) and bonds is deliberately exaggerated for clarity. (Adapted from [70])
Hydrophobic interactions of alkanethiol SAM can also be exploited to assemble a second phospholipid layer, creating a so-called hybrid bilayer membrane (HBM) [73, 74]. This system resembles a true biological membrane at least as regards the second layer and the internal hydrophobicity. In particular, the SAM turns the bilayer into a highly stable structure [75]; however, its effect on the lateral mobility of phospholipid molecules of the layer significantly limits its biomimetic properties, especially when channelling phenomena have to be observed [76]. In contrast, HBMs are well suited for investigating reactions occurring at the membrane interface such as protein surface interactions, binding of ligands to membrane-confined receptors and embedding of some redox proteins, which make these interfaces very promising for biorecognition and biosensing applications. Hydrophobic interactions have been exploited to immobilize cytochrome c oxidase in a HBM that is able to undergo direct electron transfer onto the electrode surface (Fig. 3a); this system has been successfully used for cyanide detection, owing to its inhibiting properties [77]. HBMs have also been used recently as a basis for developing multichannel biochips exploiting controlled superficial charge density (Fig. 3b) [78].

To achieve highly biomimetic properties of reconstituted systems, bilayer lipid membranes (BLMs) anchored to the substrate by suitable tethers have been developed [79, 80]. These membranes, named tethered BLMs (t-BLMs), are reconstituted by using a membrane phospholipids solution where a relatively low number of tethered phospholipid molecules are present [79–81].
Electrostatic interactions
Studies on cytochrome c revealed that electrostatic immobilization is a relatively mild method enabling direct electron transfer between protein and electrode, and does not seem to significantly affect its structure and properties [82, 83]. The electrostatic immobilization is particularly suitable for SAMs having either positively charged amine or negatively charged carboxyl groups on the surface. Successful immobilization requires the protein be sufficiently charged and the distance between the electrode and the protein redox groups not be too long. The fulfillment of these requirements can be deduced from both the protein isoelectric point and the analysis of the most likely conformation of the protein on the SAM. Physical adsorption of 32P-labelled DNA on a carboxy-modified alkyl thiol SAM has been investigated [84]. As other important anion-terminated SAMs and based on biomimetic rationales, phosphate- and phosphonate-terminated SAMs have been studied as a functional interface to immobilize proteins [85–87]. The ionic phosphate surface exhibits lower platelet reactivity than the carboxylic control; however a structure with a lower order of packing was noted on the pure SAM with a bulky phosphate terminal end [86]. In recent reports, the direct electrochemistry of cytochrome c [88] and haemoglobin [89, 90] immobilized on phosphate-terminated SAMs was investigated. The immobilized haem proteins retain their native structure and direct electron transfer is detected; this indicates that the phosphate-terminated functional interface provides a favourable biocompatible microenvironment for the protein. The interaction between the proteins and phosphate groups is likely a hydrogen-bonding interaction rather than an electrostatic interaction [89]. These studies show that the phosphate groups play a very important role in biological studies and provide a functional interface in the fabrication of electrochemical biosensors.
As noted above, the major advantage of this immobilization is that neither additional coupling reagents nor further modification of the protein is required. However, non-covalent immobilization typically involves relatively weak and reversible interactions; this has several implications for the overall robustness and reusability of systems, particularly when they are used in analytical assay and sensor devices. During the adsorption, the proteins often undergo conformational changes and denaturation that can significantly reduce their activity [91]. Furthermore, since the packing density of the immobilized proteins is uncontrolled, their activity may be further reduced by steric hindrance and non-specific adsorption [92, 93].
The turning point in the use of model substrates was the development of so-called inert surfaces that prevent the non-specific adsorption of proteins. A particularly versatile approach for suppressing non-specific adsorption is based on alkanethiol SAMs with an oligo(ethylene glycol) (OEG) chain at the termini [94–96]; the crystalline helical and amorphous forms of these kinds of SAMs prevent protein adsorption, as demonstrated in an elegant study by Whitesides and Prime [97]. The amount of protein adsorption on gold modified with ethylene glycol and unfunctionalized alkanethiol mixed SAMs is a function of both the density of the ethylene glycol adsorbates and the length of the terminal chain [97]. Indeed, the length of this chain is crucial for binding interfacial water molecules by the ethylene glycol layer, which is very important for conferring to the SAM the ability to prevent protein adsorption [98]. The importance of water penetration into the OEG terminus of the films to render them protein-resistant was demonstrated [99]. The amount of water penetrating into the SAM of OEG depends on the lateral density; this can be regulated by controlling the adsorption of the molecules on the gold surface. Computer simulations and neutron reflectivity measurements of the SAM in contact with water revealed that the water concentration inside a defect-free gold-supported film is one molecule per OEG unit, whereas essentially no water adsorption was observed on silver-supported monolayers owing to their higher packing density [100]. Thus, entropic forces play at most a limited role in densely packed SAMs. To understand the contributions to protein resistance of OEG, it was suggested that the unfavourable change in free energy when removing the strongly bound water in the interface should prevent protein adsorption as result of attractive van der Waals interactions. The combination of high loading capacity and low non-specific adsorption of biomolecules in one surface coating would provide a “perfect” sensitive layer [101]. Therefore, a mixed OEG obtained by immobilizing OEGs of various lengths onto the surface was investigated to form a structure with high binding capacity but keeping the benefit of minimal non-specific binding [102]. However, the susceptibility of ethylene glycol chains to self-oxidation limits their long-term applications. Surface phospholipids also minimize non-specific binding; their strong hydration capacity, achieved by electrostatic interactions, is hypothesized to be responsible for this effect [103]. For example, the zwitterionic properties of oligophosphorylcholine SAMs result in the suppression of kinetically irreversible non-specific adsorption of proteins; however, phosphorylcholine monolayers are not very stable [103]. The ability of a “solute” to render surfaces resistant to the adsorption of proteins has also been studied [104]. There are elements of osmolytes (organic compounds affecting osmosis) or kosmotropes (organic compounds contributing to the stability and structure of water–water interactions) that are responsible for the surfaces resistance to the adsorption of proteins (Fig. 4). The conformational flexibility of the OEG chains may contribute to their preferential exclusion and kosmotropicity, and to the protein resistance of surfaces functionalized with OEG [105–108]; conformational flexibility is, however, not a prerequisite for kosmotropicity of a solute, and is also not required for protein resistance of a surface, albeit it may sometimes be important [103]. These elaborate approaches proved that when elements of certain osmolytes and kosmotropes were incorporated into alkanethiolates such as betaine, taurine and hexamethylphosphoramide, SAMs of these compounds displayed improved protein repellency [109, 110].

Protein-resistant surfaces: a solutes preferentially excluded from the near-surface solution domain of a protein cause substantial preferential hydration of the protein; b attaching such a solute to a surface minimizes (unspecific) protein adsorption. (From [103])
Covalent interactions
For more stable attachment, the formation of covalent bonds is required; in general, proteins have many functional groups, mainly in the amino acid side chains that are suitable for immobilization purposes. As non-covalent adsorption, these methods can be used on unmodified proteins since they rely only on naturally present functional groups. A number of different classes of organic reactions have been explored for modifying the surfaces of SAMs, including nucleophilic substitutions, esterification, acylation and nucleophilic addition [111]. Under appropriate experimental conditions, the terminal groups exposed on the surface of a SAM immersed in a solution of ligands can react directly with the molecules present in solution. SAMs with maleimide functional groups react with proteins having an accessible cysteine residue [112]; the maleimide groups strongly favour conjugate addition with thiols at physiological pH (6.5–7.5), since under these conditions amines are predominantly protonated. The disulfide–thiol exchange process appears to occur more readily than displacement of the thiol on the surface; such a method is widely used to attach thiol-modified peptides and proteins to SAMs [113, 114]. The steric bulk of the thiol-modified biomolecules may hinder their transport into defect sites on the surface. The ligand immobilization on SAMs by reaction with primary amines, thiol and o-phthaldialdehyde has recently been described [115]. These methods enable the immobilization of proteins through amino coupling using a chemistry which is an alternative to peptide bond formation; the amino coupling is based on the reaction of thioacetales, formed by reaction of thiol groups with aldehydes, with primary amino groups, without any preactivation of the surface, making it suitable for sensor/array fabrication.
By far the most common method to covalently attach proteins to surfaces involves first the functionalization of the SAM surfaces with a reactive intermediate, which is then coupled to a ligand (Fig. 5). In this strategy, the common intermediate can react with a variety of ligands and it allows, in principle, spatial discrimination of active and inactive regions; so, the reactivity of regions on the surface can be turn on and off. One of the experimentally simplest and most broadly applicable methods developed for modifying SAMs exploits the formation of amide linkages via an interchain anhydride intermediate [116, 117]; trifluoroacetic anhydride dehydrates the terminated carboxylic acid of a SAM, which exposed to amine groups of the lysine chain of the protein generates the amide bonds. As an alternative, aldehyde groups can be coupled with exposed amines on proteins to produce an imine that can be reduced by sodium cyanoborohydride, or an equivalent reagent, to form a stable secondary amine linkage [118, 119]. The nucleophilicity of the amine group also allows reaction with epoxide-functionalized materials [120]. Epoxides have the advantage of being relatively stable to hydrolysis at neutral pH, which allows easy handling of the materials but can result in slow or incomplete coupling. It is important to note that the high abundance of amino groups of the lysine side chain (greater than 10%) can lead to protein attachment through many residues simultaneously, thereby restricting the degrees of conformational freedom and also increasing heterogeneity in the population of immobilized proteins. The aforementioned methods do not facilitate site-specific immobilization, except in unusual cases such as light harvesting complex 2 from Rhodobacter sphaeroides, in which the lysine residues are all found on the cytoplasmic face of the protein, which means that active ester methods yield immobilization with controlled orientation [120]. In this example, alkyl thiols in SAMs were selectively converted into weakly bound alkyl sulfonates using a near-field scanning optical microscopy technique. The sulfonates were then replaced by carboxylated alkyl thiols activated with carbonyldiimidazole. Subsequent incubation with light harvesting complex 2 leads to nanopatterns of less than 100 nm. However, the active ester method has nevertheless been widely used.

Methods used for non-specific covalent immobilization with nucleophilic residues of proteins. The reaction of lysine residues with N-hydroxysuccinimide (NHS) ester (a) or aldehydes (b) and cysteine residue bonding to maleimide groups (c) or disulfide-derivatized surfaces (d)
The aspartate and glutamate residues can be also used for immobilization by converting them in situ into the corresponding active esters with a carbodiimide coupling agent and an auxiliary nucleophile (Fig. 6). The most commonly used example of the former is N-ethyl-N-(3-dimethylaminopropyl) carbodiimide (EDC), whereas N-hydroxysuccinimide (NHS) is widely used as the auxiliary to generate the NHS ester on the protein [121–123]. This active ester can react with an amine-bearing support. The immobilization efficiency depends on several parameters, for example pH, concentration, ionic strength and reaction time, and in some cases there is the risk that the NHS ester formed on the protein molecule may then couple to other protein molecules to give poorly defined polymers. Various chemically modified surfaces are commercially available for bioanalytical applications, and some of them are designed to suppress non-specific adsorption. Many of these methods have been established and refined during the development of surface technologies, providing robust platforms for in situ monitoring of binding events (e.g. protein–protein interactions). Surface acoustic wave (SAW) devices are important types of biosensors where the in situ mass change is used for analyte detection [124]. The SAM formation on a SAW device affects its features and properties. The formation of a siloxane SAM film on a lithium niobate substrate makes it compatible with conventional SAW device manufacturing techniques [125]. This type of mass-sensitive technique is highly useful to study monolayer formation, enzyme immobilization and selective sensing of small organic molecules or large biomolecules [126, 127].

Covalent immobilization of protein with an accessible glutamate or aspartate residue to an amine-functionalized SAM by using an N-ethyl-N-(3-dimethylaminopropyl) carbodiimide (EDC)-mediated reaction and the in situ generation of a NHS active ester
Recently, different procedures have been adopted for creating locally activated carboxylic acid groups on surfaces, allowing for the site-selective attachment of proteins. With use of reactive microcontact printing [128] and microstamping [129], spatially defined patterns of activated carboxylic acid groups were fabricated to immobilize laminin and fibronectin. Precise positioning of mercaptoaldehydes in SAMs by using an atomic force microscope allows the creation of IgG and metalloprotein patterns [130–132].
Although classic chemoligation is widely used for protein immobilization, such reactions are characterized by a number of drawbacks as a consequence of the non-specific nature of the underlying attachment chemistry. Furthermore, the properties of the immobilized proteins may be partially or even completely lost. To ensure accessibility of the protein’s active site and thereby enable the detailed study of protein functions, a homogeneous surface orientation of proteins on modified surfaces of SAMs, without affecting their conformation and function, should be sought. These methods are mainly based on non-covalent interactions rather than covalent reactions to stabilize the adsorbing biomolecules. In addition to homogeneous and oriented attachment, the reversibility of immobilization can be very attractive from an economical point of view, because chip and sensor surfaces might be recyclable and suitable for repeated use.
Chelator–His-tag interactions
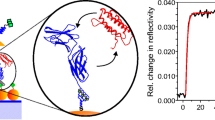
A chelator-based immobilization strategy, very often used for chromatographic protein purification, exploits the use of oligohistidine tags (“His-tags”) in combination with metal ions immobilized via chelators such as iminodiacetic acid [133] and nitrilotriacetic acid (NTA) [134, 135]. This small tag, usually consisting of six sequential histidine residues, chelates transition metals including Cu(II), Co(II), Zn(II), and Ni(II), although the latter is most commonly employed. Currently, recombinant proteins bearing an engineered His6-tag are produced by genetic engineering, thus enabling immobilization of His6-tagged fluorescent proteins, antibody, virus proteins and growth factors on Ni-NTA surfaces [136–138]. The immobilization of His6-tagged proteins on Ni-NTA SAMs, where it can be easily implemented, offers several distinct advantages over biochemical recognition elements such as antibodies systems: (1) His6-tags bound to either the C-terminus or the N-terminus are commercially available for a large number of proteins; (2) the formation of a histidine–Ni(II) complex is a fast reaction without significant alteration of the protein functionality; (3) the reaction is reversible when the surface is exposed to a stronger chelating agent, such as ethylenediaminetetraacetic acid (EDTA) or when using a competitive agent such as imidazole; (4) the spatially defined attachment of His-tags on proteins or antibodies offers the opportunity for their controlled immobilization (i.e. orientation and distance) to the support [136, 139–141]. However, the general selectivity level of this method is relatively low since several endogenous proteins have been recognized to be able to bind the metal ion, thus competing with the desired His6-fused protein [142]; the strength of the binding interaction is also relatively weak \( \left( {{K_{\rm{d}}} = {1} - {1}0\mu {\hbox{M}}} \right) \), potentially leading to unwanted dissociation of immobilized protein. A promising strategy, in this regard, is based on the use of NTA-derivative-based multivalent chelators that has been proven to be powerful and superior in comparison with monovalent NTA: the binding affinity of the His6-tag for NTA receptors increases by several order of magnitude [138, 143–145]. A systematic structural and functional study of SAMs formed on gold from a series of alkyl thiol compounds containing mono-, bis-, and tris-NTA moieties (Fig. 7) with various ratios of a protein-repellent matrix thiol, like tri(ethylene glycol)-terminated alkyl thiol, was performed by the Tampé group [143, 144]. The functional characteristics of mono-, bis- and tris-NTA were studied by SPR; the immobilization of ifnar2-His6 and its interaction with the ligand IFNα2 on 3 and 30 mol% mono-, bis- and tris-NTA SAMs revealed an increased stability of the His-tagged proteins with increasing multivalency of the chelator ligand as reported in Fig. 8. In particular, the tris-NTA SAM displayed an extraordinary stability of the ifnar2-His6 receptor even at very low surface coverage (3 mol%). The high excess of Ni(II) ions on the surface, which is required to achieve efficient immobilization through mono-NTA, strongly increases non-specific binding of proteins [145]. Thus, the use of tris-NTA low-density SAMs providing a more selective capture of proteins and detection of binding partners is very promising in functional proteomic applications.

Space-filling models and chemical structures of the filling compound tri(ethylene glycol)-terminated alkyl thiol (EG 3 ) and the mono- and multivalent chelator derivatives used to form SAMs on gold. NTA nitrilotriacetic acid. (From [145])

Functional demonstration of the multivalent chelator chips with surface plasmon resonance (SPR). Sensorgrams showing immobilization of 500 nM ifnar2-His6 (1), followed by injection of 100 nM IFNα2 (2) and 1 mM imidazole (3) on surfaces of 30 mol% (top graph) and 3 mol% (middle graph) mono-NTA (black line), bis-NTA (red line) and tris-NTA SAM (blue line). As a control experiment, the same sequence of samples were injected on the surfaces after removal of NTA-chelated Ni(II) ions with 200 mM ethylenediaminetetraacetic acid (bottom graph). Note the different scaling of the y-axes. (From [145])
The SAMs designed to achieve controlled immobilization of His6-tagged proteins have been characterized (composition, structure and order) with complementary surface analytical techniques [146]. The backfill of the OEG diluting thiol into the NTA films improved the orientation of NTA head groups and increased the order within the hydrocarbon region of the monolayer. SPR measurements showed that the maximum specific binding with a slow dissociation rate of His6-tagged proteins was achieved on the highly packed pure NTA monolayer, after which the amount of specific binding decreased owing to significant OEG dilution of the NTA head groups.
In a recent report, the interaction of the extracellular domain of transmembrane proteins N-cadherin and L1, a member of the immunoglobulin superfamily, with NTA-terminated SAMs was studied [147]. Spectroscopy investigation revealed that L1 interacts by replacing capping ethanol molecules, which are most likely linked to the carboxylate groups of the NTA complex (Fig. 9). Although cell biology experiments indicate the directional complexation of L1, these recent findings suggest that the α-helices and β-sheets of L1 lack any preferential ordering. Recently, Marin et al. [148] immobilized His6-tagged rhodopsin proteins onto SAMs; mass spectrometry was employed to carry out functional assay based on rhodopsin photoactivation. Retinal is the chromophore of rhodopsin; it is a polyene molecule with four double bonds that can have cis and trans configurations. 11-cis-Retinal, in contrast to the trans isomer, combines with the opsin apoprotein, via Schiff-base linkage, to form rhodopsin. Light transforms 11-cis-retinal of rhodopsin into the trans isomer. Photoisomerization of 11-cis-retinal leads to a conformational change allowing the binding of the transduction protein complex. Moreover, interaction with non-hydrolysable nucleotides blocks the interaction of transduction with rhodopsin.

The chemical structure of the surface NTA complex before (left) and after (right) the binding of L1 to the NTA complex. Upon binding, the ethanol molecules that cover the carboxylate groups are displaced by the protein. (From [147])
The interaction of a Ni-NTA-terminated SAM with a His-tag has also been exploited for reconstruction of BLMs. These membranes, named t-BLMs [79, 80], are assembled by fusion of liposomes embedding His-tagged transmembrane protein on a Ni-NTA-terminated mixed SAM. As mentioned above, the relative amount of tether molecules is crucial to get the best performance; in particular, it should not exceed 10 mol% [149] and as a diluting component of the mixed SAM a quite short OH-terminated thiols, creating the necessary hydrophilic environment, are usually employed [79]. The resulting BLM embeds the transmembrane protein, which is blocked onto the surface-anchored Ni-NTA derivative through the interaction of its His-tag. The presence of tethers confers stability to the BLM, which is also characterized by good biomimetic properties, thanks to the hydrophilic aqueous reservoir present between the support and the BLM [150, 151]. This approach has been adopted to obtain a BLM embedding cytocrome c oxidase tethered through a His-tag to a Ni-NTA-functionalized SAM [152, 153]. In this case the His-tag was attached to subunit II of cytochrome c oxidase molecules that were assembled with their binding site facing the substrate, thus allowing a very efficient electron transfer from the electrode to the redox centre (Fig. 10).

Tethering membranes via incorporated proteins: the solubilized membrane proteins are covalently attached to the gold electrode, e.g. via NTA/Ni2+ coordination to the His-tags of a genetically modified cytocrome c oxidase. The lipid bilayer is assembled around the protein by in situ dialysis of micelles made from detergent-solubilized lipid. (From [80])
A key application that has to be considered is the spatial distribution and orientation of redox proteins on the surface, which has been predicted to play a significant role in the electrochemistry of the redox protein monolayer, because the redox-active site is generally not located at the centre of the protein [154, 155]. As a result, a distribution of molecular orientations should generate a distribution of redox-active site–electrode separation distances and hence a distribution of electron transfer rate constants and redox potentials [155]. Among the few protein-immobilization strategies able to satisfy these multiple criteria, the method based on SAMs of Ni-NTA/His-tag technology is the most suitable. Accordingly, the mediated electrochemistry of His-tagged ferredoxin/NADP + reductase [156], the haemoprotein human neuroglobin [157] and laccase [158], specifically bound to NTA-modified electrodes, was reported (Fig. 11). From the quantitative analysis of the catalytic current recorded at these enzyme electrodes, corroborated by an independent measurement of the active-enzyme surface concentrations, it was shown that the activity of the enzyme was fully preserved, indicating that NTA coatings provide a biocompatible environment for preserving the functionality of bound proteins. Nevertheless, whatever the redox enzymes investigated, no enzymatic activities could be observed without the participation of a soluble mediator. The lack of a direct electron transfer could be ascribed to the high distances between the electrode and the active site of these enzymes. Another facilitated direct electron transfer between a gold electrode and His-tagged proteins was proposed at a triazacyclonane ligand, an alternative chelator complex, covalently attached to a short-length SAM on gold [159]. From the distinct reversible cyclic voltammetry curves of several His-tagged redox proteins, the authors concluded that a rapid electronic communication occurs between the protein redox sites and the electrode. However, the apparent standard potentials of the immobilized proteins did not agree with those obtained in solution; in addition, the possible structural changes and/or protein denaturation on this very short SAM were not taken into consideration, preventing any comment regarding the protein integrity.

Two-step synthesis of the thiol–NTA ligand from thioctic acid (1, 2), thiol chemisorption on the gold surface (3), pretreatment of the NTA-modified surface with an aqueous solution of CuCl2 (4) and addition of the His-tagged protein to the modified surface (5). (From [158])
Supramolecular interactions
Another strategy for modifying the surface composition, through non-covalent interactions providing the highest degree of specificity, is based on the use of suitable designed supramolecular interactions [160]. Monolayers based on supramolecular host–guest interactions of cyclodextrin SAMs, also called “molecular printboards”, offer unique properties for immobilizing proteins through small multivalent, orthogonal linker molecules [161]. This method allows control both over the binding strength, by varying the valency of the linker at the printboard, and over the orientation of the protein, by the bioengineering of a specific binding site at a predetermined location in the protein. These monolayers have recently been applied to study the specific binding of some model proteins [162–164]; the multiple “host–guest” interactions can stabilize the non-covalent assembly, and their selectivity suggests that, in principle, it is possible to place two ligands very close together on a surface.
Bioaffinity interactions
The control over protein adsorption at surfaces and interfaces is also one of the current challenges for mimicking biological interfaces, proteomics and medicine screening. In most cases, this control relies upon a “key–lock system” for the specific attachment of a particular protein to an engineered surface. Biochemical affinity reactions allow a gentle oriented immobilization of proteins, providing an important advantage over other orienting immobilization techniques. Additionally to the realization of an oriented and homogeneous attachment, by this method it is also possible to detach proteins and to use the same surface more times. Undoubtedly, one of the most well-known and extensively researched protein-mediated immobilization methods exploits the non-covalent interaction of either avidin (or alternatively streptavidin (SAv)) with biotin-functionalized proteins [165–167]. Avidin is a homotetrameric protein with individual subunits, every one of which can bind to biotin (vitamin B7/H) with a high degree of specificity and affinity (K D = 10-15 M), thereby making the avidin–biotin system one of the strongest known non-covalent binding entities in nature [167, 168]. In pioneering experiments, Häussling et al. [169] and Müller et al. [170] showed that immobilization of SAv could be performed on a specifically designed SAM [169, 170]. Keeping in mind both specific and non-specific adsorption of SAv, the possibility to use mixed SAMs of biotin-substituted and hydroxyl-substituted alkanethiols assembled on gold was successfully explored; in the early 1990s it was shown that they provided a platform for the fabrication of novel biosensors [169–171]. A more detailed study on these mixed SAMs to optimize the immobilization with respect to surface coverage, specificity and activity for various types of SAv (both wild type and mutants) has been carried out [172]. Recently, it was found that biotinylated reductase, immobilized onto a biotin–SAv chip in a uniformly oriented way, showed a remarkable increase of activity in comparison with the randomly immobilized enzyme [173]. Different biotinylated protein A/G were immobilized on parallel channels for use in sensor applications [174], whereas the use of a bifunctional linker, such as 2-(biotinamido)ethanethiol, enabled the preparation of immunoarrays on gold surfaces [175].
In another biosensor set-up, the model enzyme β-lactamase was genetically engineered to insert a single cysteine at a specific site of its surface; then the residue was biotinylated with the aim of obtaining an enzyme molecule immobilized with the right orientation especially as regards its active site [176]. A binary mixture of biotin-terminated thiols and hydroxyl-terminated diluting thiols was assembled onto a gold surface to form a biotinylated SAM as shown in Fig. 12. Once the binding of the NeutrAvidin monolayer with molecularly controlled orientation had been established, the remaining free binding sites exposed to the aqueous phase were used to immobilize the biotinylated enzyme. By SPR measurements it was demonstrated that surfaces with different coverage degrees of the enzyme can be obtained by adding differently concentrated biotinylated β-lactamase to the NeutrAvidin monolayer coated surfaces.

Lactamase chip set-up: different biotinylated β-lactamase solution concentrations were immobilized through a biotin–NeutrAvidin–biotin sandwich on a SAM created on a gold surface, resulting in different enzyme surface coverage. The chip was then used to monitor enzymatic activity of the immobilized enzyme by SPR. (Adapted from [176])
The biotin–avidin interaction has recently been used for functionalizing a HBM [177]. A suitable avidin-functionalized protein was incubated with a biotinylated octadecanethiol/phosphatidylcholine HBM, providing a convenient tool for the preparation of sensor surfaces. The proposed method, investigated and optimized for the realization of a QCM sensor chip, was characterized by enhanced performance compared with the analogous system directly anchored by the EDC/NHS method: furthermore, the polar head groups of phosphatidylcholine on the sensor surface significantly reduced the protein non-specific adsorption. Analogously, Elie-Caille et al. [178] used biotin–avidin interaction to prepare t-BLM for studying mitochondrial proteins embedded into the membrane. Figure 13 illustrates the procedure for t-BLM preparation, whose behaviour and performance were studied by SPR, fluorescence and fluorescence recovery after photobleaching.

The step-by-step assembly for the supported membrane. Whatever the solid support, gold or glass, the process starts from the same amine monolayer. Note that the formation of the continuous bilayer is triggered by a concentrated solution of PEG (molecular weight 8,000) in the last step. (From [178])
A critical point of the biotin–SAv approach for creating proteins patterns is the optimization of the protein–target interaction: probe–target binding at highly packed probe arrays may differ significantly from the same binding occurring in a biological setting [179]. These differences can be caused by several probe–probe interactions (e.g. steric or electrostatic interactions between probe molecules) that prevent binding of the target. For generation of probe surfaces, an optimal method is one allowing control over the space around each probe individually to both reduce probe–probe interactions and maximize the density of probe molecules (and target binding capacity as well); these attributes are desirable for applications that require highly sensitive sensing or arrayed interfaces.
Significant research efforts have been addressed for the fabrication of mixed SAMs comprising various types of biotin-substituted alkanethiols and other alkanethiols to study different aspects of avidin immobilization, and its variants, on the engineered molecular templates for biosensor applications. A biotinylated surface, whose biotin probe density was suitably controlled by using a labile dendron spacer, was recently reported [180]. In this process, anchor molecules are first adsorbed to a surface, with dendron modifiers attached; steric interactions of the bulky dendrons control the density of anchor molecules bound to the surface. The dendron branches are subsequently detached from the anchor molecules, and the anchors are chemically modified with probe (biotin) molecules; this produces a surface with controlled spacing among probe molecules (Fig. 14).

An immobilization method that controls the space around individual probe molecules: 1 fabrication of a dendrimer monolayer, 2 removal of dendron spacers and introduction of a matrix molecule and 3 modification of a probe molecule. (From [180])
A method for protein patterning based on an electrochemically active biotin derivative that generates a bioactive biotin surface by a mild electrochemical perturbation has been described [181]. In this case a hydroquinone protecting group is electrochemically oxidized to benzoquinonium cation; subsequently, benzoquinone and CO2 are released after nucleophilic acyl substitution by water and, finally, the bioactive biotin surface is generated (Fig. 15). Since this method can be conducted under neutral buffered conditions in a short time, it enables the serial patterning of multiple proteins, whereas previously attached proteins retain their activity. In a photochemistry approach, deactivated biotin contains a photolabile protecting group that can be selectively activated by photoirradiation to obtain patterns of biotin available for binding proteins. For example, when all amine groups in SAMs are initially masked with methyl-6-nitroveratryloxycarbonyl groups, local release induced by photolysis allows for the incorporation of biotin [182, 183]. This technique, however, requires the synthesis of alkanethiols containing photoprotecting groups and the availability of a photomask (Fig. 16). The ability to produce multiple, aligned patterns of SAMs in a single step, without alignment of photomasks in separate steps, increases the versatility of SAMs for studying different interfacial phenomena, including dewetting and adhesion.

The electrochemically oxidative reaction for the generation of bioactive biotin on the hydroquinone-caged biotin-modified gold surface. (Adapted from [181])

Preparation and immunolabelling of multiple, aligned SAMs. a Patterning multiple, aligned SAMs using a photomask. A mixed SAM containing HS(CH2)11EG2NPOC and HS(CH2)11EG6OH was illuminated through an area-selective mask that transmitted light at either 220 or 365 nm only or that blocked light at all wavelengths, to produce a region containing the original SAM, a SAM that terminated in primary amines, and a region of bare gold (or oxidized gold). This allowed (+)-biotin NHS ester to react with the primary amines and also formed a new SAM composed of HS(CH2)11EG2DNP and HS(CH2)11EG6OH on the exposed gold. The SAM was labelled using anti-biotin mouse IgG (followed by fluorescently labelled anti-mouse IgG) and anti-DNP rabbit IgG (followed by fluorescently labelled anti-rabbit IgG). b Fluorescence images of patterns of multiple, aligned SAMs. (From [182])
A new photochemical method for the site-specific immobilization and patterning of proteins employing a thiol–ene reaction onto a surface by using the biotin–SAv approach has also been developed [184]. An olefin-modified biotin derivate was photochemically attached to a thiol-functionalized surface; a SAv-patterned surface was produced by Cy5-labelled SAv incubation. The SAv patterns were used as a template for alkaline phosphatase and Ras GTPase immobilization; this template allows the retention of structure and activity of considered proteins.
Recently, Ballav et al. [185–188] developed an alternative approach, based on irradiation-promoted exchange reaction (IPER), to prepare mixed SAMs of OEG-substituted alkanethiols and biotin-substituted alkanethiols on gold for specific attachment of target protein [188]. In this approach, the mixing of both components of a binary film occurs by the exchange reaction between the primary SAM (one component) and a potential molecular substituent (second component) (Fig. 17). Electron or UV irradiation creates subtle structural and chemical defects in the primary SAM, which promote the molecular exchange. As a result, the kinetics and extent of the exchange reaction can be tuned by selection of a proper irradiation dose, so a mixed SAM of a desired composition can be prepared. The tenability of the SAM composition is one of the major advantages of IPER in comparison with the lithographic technique exploiting UV-promoted molecular exchange [182]. In general, the avidin immobilization onto the mixed SAMs prepared by IPER is found to be consistent with earlier reports on analogous films fabricated by the coassembly method [189]. The concentration of the biotin-substituted alkanethiol component in the mixed SAMs necessary for the maximum surface coverage of the specific protein was found to be somewhat lower than the expected value; in contrast, in the case of the use of IPER, the maximum avidin coverage was somewhat higher than that obtained by means of the coassembly method. The authors ascribed these differences to the lack of phase segregation and to better separation of the individual biotin-substituted alkanethiol species in the SAM matrix in the case of IPER.

The individual steps and processes occurring during the fabrication of mixed SAMs designed for avidin immobilization by irradiation-promoted exchange reaction (IPER). The starting point is highlighted by the grey background. Step 1: general effect of electron irradiation on the oligo(ethylene glycol) (OEG) HO(CH2CH2O)3(CH2)11SH (EG3) and HO-(CH2CH2O)7(CH2)11SH (EG7) SAMs. Step 2: depending on the identity of the primary OEG SAM, the outcome of IPER with a biotin-substituted alkanethiol (BAT) can be different. Steps 2a and 2b are characteristic of the EG3/EG3-Bio and EG7/EG3-Bio systems, respectively. Step 3: adsorption of a specifically binding protein (avidin) on the mixed OEG/BAT SAMs prepared by IPER. Steps 3a and 3b are characteristic of the EG3/EG3-Bio and EG7 + EG3-Bio systems, respectively. Step 4: verification of non-specific proteins adsorption onto the mixed OEG/BAT SAMs designed for the specific protein immobilization. Steps 4a and 4b are characteristic of the EG3/EG3-Bio (no adsorption of bovine serum albumin) and EG7/EG3-Bio (some adsorption of globulin) systems, respectively. Step 5: non-specific adsorption of proteins (including avidin) onto the irradiated OEG SAMs. (From [188])
Concluding remarks
SAMs of alkanethiols on gold are prototypal surfaces for the fabrication of highly efficient protein biochips. The flexibility to design different head groups of monolayers using a large number of functional groups makes this functionalization strategy especially useful for the controlled fabrication of structurally ordered assemblies of proteins on surfaces. SAMs are also a very useful tool to adjust surface properties: a lot of methods have been established to obtain chemical gradients. The formation of mixed a thiol–OEG monolayer generates a surface displaying protein resistance and adequate surface coverage without creating steric hindrance among immobilized molecules. The past few years have witnessed impressive progress in the design of different immobilization strategies attempting to obtain full coverage of the surface and the best detection performance. Furthermore, SAMs of alkanethiols on gold have been highlighted as one of the most promising surfaces owing to the ease of real-time detection via imaging techniques.
Although the details of the thermodynamics, kinetics and mechanisms of assembly will differ significantly, the recent progress of SAM technology in obtaining full control over nanofabrication indicates that the next generation of functional protein chips possessing unique architecture through molecular design will soon be available for use as biochips and devices.
References
Phizicky E, Bastiaens PIH, Zhu H, Snyder M, Fields S (2003) Nature 422:208–215
Wilson DS, Nock S (2003) Angew Chem Int Ed 42:494–500
Bilitewski U (2006) Anal Chim Acta 568:232–247
Zheng G, Patolsky F, Cui Y, Wang WU, Lieber CM (2005) Nat Biotechnol 10:1294–1301
LaBaer J, Ramachandran N (2005) Curr Opin Chem Biol 9:14–19
Kingsmore FS (2006) Nat Rev Drug Discov 5:310–320
Lueking A, Cahill DJ, Müllner S (2005) Drug Discov Today 10:789–794
Lee JH, Wark AW, Corn RM (2008) Analyst 133:975–983
Weinrich D, Jonkheijm P, Niemeyer CM, Waldmann H (2009) Angew Chem Int Ed 48:7744–7751
Rusmini F, Zhong Z, Feijen J (2007) Biomacromolecules 8:1775–1789
Wong LW, Khan F, Micklefield J (2009) Chem Rev 109:4025–4053
Jonkheijm P, Weinrich D, Schröder H, Niemeyer CM, Waldmann H (2008) Angew Chem Int Ed 47:9618–9647
Sarma AK, Vatsyayan P, Goswami P, Minteer SD (2009) Biosens Bioelectron 24:2113–2322
Mrksich M (2005) MRS Bull 30:180–184
Niemeyer CM, Mirkin CA (eds) Nanobiotechnology, vols I and II, Wiley-VCH, Weinheim, 2004, 2007
Ulman A (1991) An introduction to ultrathin organic films from Langmuir–Blodgett to self-assembly. Academic, New York
Ball P (1994) Designing the molecular world. Princeton University Press, Princeton
Haick H, Cahen D (2008) Prog Surf Sci 83:217–261
Flink S, Van Veggel FCJM, Reinhoudt DN (2000) Adv Mater 12:1315–1328
Ulman A (1996) Chem Rev 96:1533–1554
Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM (2005) Chem Rev 105:1103–1169
Davis F, Higson SPJ (2005) Biosens Bioelectron 21:1–20
Love JC, Wolfe DB, Haasch R, Chabinyc ML, Paul KE, Whitesides GM, Nuzzo RG (2003) J Am Chem Soc 125:2597–2604
Stapleton JJ, Daniel TA, Uppili S, Cabarcos OM, Naciri J, Shashidhar R, Allara DL (2005) Langmuir 21:11061–11069
Amato C, Devillers S, Calas J, Delhalle J, Mekhalif Z (2008) Langmuir 24:10879–10885
Thompson WR, Pemberton JE (1995) Langmuir 11:1720–1731
Liu H-B, Venkataraman NV, Spencer ND, Textor M, Xiao S-J (2008) Chemphyschem 9:1979–1981
Raynor JE, Petrie TA, García AJ, Collard DM (2007) Adv Mater 19:1724–1728
Neves BRA, Salmon ME, Russell PE, Troughton EBJ (2001) Langmuir 17:8193–8198
Flink S, van Veggel FCJM, Reihould DN (2001) J Phys Org Chem 14:407–415
Hou Z, Abbott NL, Stroeve P (1998) Langmuir 14:3287–3294
Mrksich M (2009) Acta Biomater 5:832–841
Mrksich M, Whitesides GM (1996) Annu Rev Biophys Biomol Struct 25:55–78
Liu D, Xie Y, Shao H, Jiang X (2009) Angew Chem Int Ed 48:4406–4408
Dubois LH, Nuzzo RG (1992) Annu Rev Phys Chem 43:437–463
Li Y, Yuan B, Ji H, Han D, Chen S, Tian F, Jian X (2007) Angew Chem Int Ed 46:1094–1096
Zhu XY, Mills KL, Peters PR, Bahng JH, Liu EH, Shim J, Naruse K, Csete ME, Thouless MD, Takayama S (2005) Nat Mater 4:403–406
Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL (2005) Nat Meth 2:599–605
Schreiber F (2000) Prog Surf Sci 65:151–256
Yang G, Liu G-Y (2003) J Phys Chem B 107:8746–8759
Mullen K, Rabe JP (2008) Acc Chem Res 41:511–520
Torrelles X, Vericat C, Vela ME, Fonticelli MH, Millone MAD, Felici R, Lee T-L, Zegenhagen J, Munoz G, Martin-Gago JA, Salvarezza RC (2006) J Phys Chem B 110:5586–5594
Li B, Zeng CG, Li QX, Wang B, Yuan LF, Wang HQ, Yang JL, Hou JG, Zhu QS (2003) J Phys Chem B 107:972–984
Dai J, Li Z, Jin J, Shi Y, Cheng J, Kong J, Bi S (2009) Biosens Bioelectron 24:1074–1082
Zhu ZH, Daniel TA, Maitani M, Cabarcos OM, Allara DL, Winograd N (2006) J Am Chem Soc 128:13710–13719
Monne J, Diez Y, Puy J, Galceran J, Nelson A (2007) Langmuir 23:10581–10588
Biebuyck HA, Bain CD, Whitesides GM (1994) Langmuir 10:1825–1833
Noh J, Murase T, Nakajima K, Lee H, Hara M (2000) J Phys Chem B 104:7411–7418
Jung C, Dannenberger O, Xu Y, Buck M, Grunze M (1998) Langmuir 14:1103–1107
Lee H, He Z, Hussey CL, Mattern DL (1998) Chem Mater 10:4148–4153
Takiguchi H, Sato K, Ishida T, Abe K, Yase K, Tamada K (2000) Langmuir 16:1703–1710
Beulen MW, Husman B-H, van der Heijden PA, van Veggel FCJM, Simons MG, Biemond EMEF, de Lange PJ, Reinhouldt DN (1996) Langmuir 12:6170–6172
Noh J, Nakamura F, Kim J, Lee H, Hara M (2002) Mol Cryst Liq Cryst 377:165–175
Noh J, Murase T, Nakajima K, Lee H, Hara M (2000) J Phys Chem B 104:7411–7416
Nuzzo RG, Zegarski BR, Dubois LH (1987) J Am Chem Soc 109:733–740
Aslam M, Bandyopadhyay K, Vijayamohanan K, Lakshminarayanan V (2001) J Colloid Interface Sci 234:410–417
Frederix F, Bonroy K, Reekmans G, Laureyn W, Campitelli A, Abramov MA, Dehaen W, Maes G (2004) J Biochem Biophys Meth 58:67–74
Arya SK, Solanki PR, Datta M, Malhotra BD (2009) Biosens Bioelectron 24:2810–2817
Mizutani F (2008) Sens Actuators B 130:12–20
Faull JD, Gupta VK (2002) Langmuir 18:6584–6592
Liebau M, Janssen HM, Inoue K, Shinkai S, Huskens J, Sijbesma RP, Meijer EW, Reinhouldt DN (2002) Langmuir 18:674–682
Ostuni E, Grzybowski BA, Mrksich M, Roberts CS, Whitesides GM (2003) Langmuir 19:1861–1872
Yue H, Khoshtariya D, Waldeck DH, Grochol J, Hildebrandt P, Murgida DH (2006) J Phys Chem B 110:19906–19913
Davis KL, Drews BJ, Yue H, Waldeck DH, Knorr K, Clark RA (2008) J Phys Chem C 112:6571–6576
Bonroy K, Frederix F, Reekmans G, Dewolf E, de Palma R, Borghs G, Declerck P, Goddeeris B (2006) J Immunol Meth 312:167–178
Nakamura F, Ito E, Hayashi T, Hara M (2006) Colloids Surf A 485:284–285
Subramanian A, Irudayaraj J, Ryan T (2006) Sens Actuators B 114:192–198
Gobi KV, Iwasaka H, Miura N (2007) Biosens Bioelectron 22:1382–1389
Fragoso A, Laboria N, Latta D, O’Sullivan CK (2008) Anal Chem 80:2556–2563
Chi Q, Zhang J, Andersen JET, Ulstrup J (2001) J Phys Chem B 105:4669–4679
Murgida DH, Hildebrandt P (2004) Phys Chem Chem Phys 7:3773–3784
Fleming BD, Praporski S, Bond AM, Martin LL (2008) Langmuir 24:323–327
Siepman JI, Mcdonald IR (1998) Thin Film 24:225–226
Plant AL (1999) Langmuir 15:5128–5135
Silin VI, Wieder H, Woodward JT, Valincius G, Offenhausser A, Plant AL (2002) J Am Chem Soc 124:14676–14683
Cornell BA, Braach-Maksvytis VLB, King LG, Osman PDJ, Raguse B, Wieczorek L, Pace RJ (1997) Nature 387:580–583
Su L, Kelly JB, Hawkridge FM, Rhoten MC, Baskin SI (2005) J Electroanal Chem 581:241–248
Suraniti E, Tumolo T, Baptista MS, Livache T, Calemczuk R (2007) Langmuir 23:6835–6842
Rossi C, Chopineau J (2007) Eur Biophys J 36:955–965
Knoll W, Köper I, Nauman R, Sinner E-K (2008) Electrochim Acta 53:6680–6689
Lang H, Duschl C, Vogel H (1994) Langmuir 10:197–210
de Groot MT, Evers TH, Merkx M, Koper MTM (2007) Langmuir 23:729–736
Xu J, Bowden EF (2006) J Am Chem Soc 128:6813–6822
Wrobel N, Deininger W, Hegemann P, Mirsky VM (2003) Colloids Surf B 32:157–162
Östblom M, Ekeroth J, Konradsson P, Liedberg B (2006) J Phys Chem B 110:1695–1702
Tsai M-Y, Sun Y-T, Lin J-C (2007) J Colloid Interface Sci 308:474–484
Chen Y, Wang F-B, Gou L-R, Zheng L-M, Xia X-H (2009) J Phys Chem C 113:3746–3750
Chen Y, Yang X-J, Guo L-R, Jin B, Xia X-H, Zheng L-M (2009) Talanta 78:248–252
Chen Y, Bo J, Guo L-R, Yang X-J, Chen W, Gu G, Xia X-H, Zheng L-M (2008) Chem Eur J 14:10727–10734
Chen Y, Yang X-J, Guo L-R, Li J, Xia X-H, Zheng L-M (2009) Anal Chim Acta 644:83–89
Gray JJ (2004) Curr Opin Struct Biol 14:110–132
Khan F, He M, Taussig MJ (2006) Anal Chem 78:3072–3079
Guo A, Zhu X-Y (2007) In: Predki PF (ed) Functional protein microarrays in drug discovery, 1st edn. CRC, Boca Raton, pp 53–71
Nelson CM, Raghavan S, Tan JL, Chen CS (2003) Langmuir 19:1493–1499
Capadona R, Collard DM, García AJ (2003) Langmuir 19:1847–1852
Petrie TA, Capadona JR, Reyes CD, García AJ (2006) Biomaterials 27:5459–5470
Prime KL, Whitesides GM (1993) J Am Chem Soc 115:10714–10721
Chen S, Zheng J, Li L, Jiang S (2005) J Am Chem Soc 127:14473–14478
Herrwerth S, Eck W, Reinhardt S, Grunze M (2003) J Am Chem Soc 125:9359–9366
Schwendel D, Hayashi T, Dahint R, Pertsin AJ, Grunze M, Steitz R, Schreiber F (2003) Langmuir 19:2284–2293
Frasconi M, Mazzarino M, Botrè F, Mazzei F (2009) Anal Bioanal Chem 394:2151–2159
Mehne J, Markovic G, Pröll F, Schweizer N, Zorn S, Schreiber F, Gauglitz G (2008) Anal Bioanal Chem 391:1783–1791
Kane RS, Deschatelets P, Whitesides GM (2003) Langmuir 19:2388–2391
Ostuni E, Chapman RG, Holmlin RE, Takayama S, Whitesides GM (2001) Langmuir 17:5605–5620
Timasheff SN (1998) Adv Protein Chem 51:355–432
Zwahalen M, Herrwerth S, Eck W, Reinhardt S, Grunze M, Hähner G (2003) Langmuir 19:9305–9310
Harder P, Grunze M, Dahint R, Whithesides GM, Laibinis PE (1998) J Phys Chem B 102:426–436
Wang RLC, Creuzer HJ, Grunze M (1997) J Phys Chem B 101:9767–9773
Skoda MWA, Jacobs RMJ, Willis J, Schreiber F (2007) Langmuir 23:970–974
Skoda MWA, Schreiber F, Jacobs RMJ, Webster JRP, Wolff M, Dahint R, Schwendel D, Grunze M (2009) Langmuir 25:4056–4064
Sullivan TP, Huck WTS (2003) Eur J Org Chem 17–29
Houseman BT, Gawalt ES, Mrksich M (2003) Langmuir 19:1522–1529
Wagner GJ, Lee HJ, Corn RM (2002) Anal Chem 74:5161–5168
Smith EA, Thomas WD, Kiessling LL, Corn RM (2003) J Am Chem Soc 125:6140–6148
Kyprianou D, Guerreiro AR, Nirschl M, Chianella I, Subrahmanyam S, Turner APF, Piletsky S (2010) Biosens Bioelectron 25:1049–1055
Ducker RE, Montague MT, Leggett GJ (2008) Biointerphases 3:59–65
Sun Y, Yan F, Yang W, Sun C (2006) Biomaterials 27:4042–4049
Hahn CD, Leitner C, Weinbrenner T, Schlapak R, Tinazli A, Tampe R, Lackner B, Steindl C, Hinterdorfer P, Gruber HJ, Holzl M (2007) Bioconjug Chem 18:247–253
MacBeath G, Schreiber SL (2000) Science 289:1760–1764
Reynolds NP, Janusz S, Escalante-Marun M, Timney J, Ducker RE, Olsen JD, Otto C, Subramaniam V, Leggett GJ, Hunter CN (2007) J Am Chem Soc 129:14625–14631
Liu J, Row MNP, Gooding JJ (2006) Chem Phys 324:226–235
Subramanian A, Irudayaraj J, Ryan T (2006) Biosens Bioelectron 21:998–1006
Yam CM, Deluge M, Tang D, Kumar A, Cai C (2006) J Colloid Interface Sci 296:118–130
Gizeli E (2000) Anal Chem 72:5967–5972
Nihonyanagi S, Eftekhari-Bafrooei A, Hines J, Borguet E (2008) Langmuir 24:5161–5165
Su WC, Zhang WG, Zhang S, Fana J, Yin X, Luo ML, Ng SC (2009) Biosens Bioelectron 25:488–492
Park I-S, Kim D-K, Adanyi N, Varadi M, Kim N (2004) Biosens Bioelectron 19:667–674
Feng CL, Embrechts A, Bredebush I, Schnekenburger J, Domschke W, Vancso GJ, Schönherr H (2007) Adv Mater 19:286–290
Bhangale SM, Tjong V, Wu L, Yakovlev N, Moran PM (2005) Adv Mater 17:809–813
Liu G-Y, Amro NA (2002) Proc Natl Acad Sci USA 99:5165–5170
Lee K-B, Park S-J, Mirkin CA, Smith JC, Mrksich M (2002) Science 295:1702–1705
Lee SW, Oh B-K, Sanedrin RG, Salaita K, Fujigaya T, Mirkin CA (2006) Adv Mater 18:1133–1136
Porath J, Carlsson J, Olsson I, Belfrage G (1975) Nature 258:598–602
Hochuli E, Bannwarth W, Dobeli H, Gentz R, Stuber D (1988) Nat Biotechnol 6:1321–1327
Gaberc-Porekar V, Menart V (2005) Chem Eng Technol 28:1306–1311
Gizeli E, Glad J (2004) Anal Chem 76:3995–4001
Zhen G, Falconnet D, Kuennemann E, Vörös J, Spencer ND, Textor M, Zürcher S (2006) Adv Funct Mater 16:243–251
Ludden MJW, Mulder A, Schulze K, Subramaniam V, Tampé R, Huskens J (2008) Chem Eur J 14:2044–2051
Lata S, Piehler J (2005) Anal Chem 77:1096–1105
Valiokas R, Klenkar G, Tinazli A, Tampé R, Liedberg B, Piehler J (2006) Chembiochem 7:1325–1329
Gamsjaeger R, Wimmer B, Kahr H, Tinazli A, Picuric S, Lata S, Tampé R, Maulet Y, Gruber HJ, Hinterdorfer P, Romanin C (2004) Langmuir 20:5885–5890
Hengen PN (1995) Trends Biochem Sci 20:187–205
Lata S, Reichel A, Brock R, Tampé R, Piehler J (2005) J Am Chem Soc 127:10205–10215
Klenkar G, Valiokas R, Lundström I, Tinazli A, Tampé R, Piehler J, Liedberg B (2006) Anal Chem 78:3643–3650
Valiokas R, Klenkar G, Tinazli A, Reichel A, Tampé R, Piehler J, Liedberg B (2008) Langmuir 24:4959–4967
Cheng F, Gamble LJ, Castner DG (2008) Anal Chem 80:2564–2573
Fick J, Wolfram T, Belz F, Roke S (2010) Langmuir 26:1051–1056
Marin VL, Bayburt TH, Sligar SG, Mrksich M (2007) Angew Chem Int Ed 46:8796–8798
Deverall MA, Gindl E, Sinner EK, Besir H, Ruehe J, Saxton MJ, Nauman CA (2005) Biopys J 88:1875–1886
Raguse B, Braach-Maksvytis V, Cornell BA, King LG, Osman PDJ, Pace RJ, Wieczorek L (1998) Langmuir 14:648–659
Krisna G, Schulte J, Cornell BA, Pace RJ, Osman PD (1998) Langmuir 19:2294–2305
Giess F, Friedrich MG, Heberle J, Nauman RL (2004) Biophys J 87:3213–3220
Ataka K, Giess F, Knoll W, Nauman R, Haber-Pohlmeier S, Richter B, Heberle J (2004) J Am Chem Soc 126:16199–16206
Araci ZO, Runge AF, Doherty WJ, Saavedra SS (2008) J Am Chem Soc 130:1572–1573
Yue H, Waldeck DH, Petrovic J, Clark RA (2006) J Phys Chem B 110:5062–5072
Madoz-Gúrpide J, Abad JM, Fernández-Recio J, Vélez M, Vázquez L, Gómez-Moreno C, Fernández VM (2000) J Am Chem Soc 122:9808–9817
Balland V, Lecomte S, Limoges B (2009) Langmuir 25:6532–6542
Balland V, Hureau C, Cusano AM, Liu Y, Tron T, Limoges B (2008) Chem Eur J 14:7186–7192
Johnson DL, Martin LL (2005) J Am Chem Soc 127:2018–2019
Villalonga R, Cao R, Fragoso A (2007) Chem Rev 107:3088–3116
Ludden MJW, Reinhoudt DN, Huskens J (2006) Chem Soc Rev 35:1122–1134
Ludden MJW, Mulder A, Tampé R, Reinhoudt DN, Huskens J (2007) Angew Chem Int Ed 46:4104–4107
Ludden MJW, Li X, Greve J, van Amerongen A, Escalante M, Subramaniam V, Reinhoudt DN, Huskens J (2008) J Am Chem Soc 130:6964–6973
Hwang I, Baek K, Jung M, Kim Y, Park KM, Lee D-W, Selvapalam N, Kim K (2007) J Am Chem Soc 129:4170–4171
Green NM (1990) Meth Enzymol 184:51–67
Smith CL, Milea GS, Nguyen GH (2006) Top Curr Chem 261:63–90
Laitinen OH, Nordlund HR, Hytönen VP, Kulomaa MS (2007) Trends Biotechnol 25:269–281
Laitinen OH, Marttila AT, Airenne KJ, Kulik T, Livnah O, Bayer EA, Wilchek M, Kulomaa MS (2001) J Biol Chem 276:8219–8224
Häussling L, Ringsdorf H, Schmitt FJ, Knoll W (1991) Langmuir 7:1837–1840
Müller W, Ringsdorf H, Rump E, Wildburg G, Zhang X, Angermaier L, Knoll W, Liley M, Spinke J (1993) Science 262:1706–1708
Spinke J, Liley M, Guder H-J, Angermaier L, Knoll W (1993) Langmuir 9:1821–1825
Pérez-Luna VH, O’Brien MJ, Opperman KA, Hampton PD, Stayton PS, Klumb L, Lopez GP (1999) J Am Chem Soc 121:6469–6478
Holland-Nell K, Beck-Sickinger AG (2007) Chembiochem 8:1071–1076
Ludden MJW, Ling XY, Gang T, Bula WP, Gardeniers HJGE, Reinhoudt DN, Huskens J (2008) Chem Eur J 14:136–142
Jung S-H, Son H-Y, Yuk JS, Jung JW, Kim KH, Lee C-H, Hwang H, Ha K-S (2006) Colloids Surface B 47:107–111
Xu F, Zhen G, Yu F, Kuennemann E, Textor M, Knoll W (2005) J Am Chem Soc 127:13084–13085
Mun S, Choi S-J (2009) Biosens Bioelectron 24:2522–2527
Elie-Caille C, Fliniaux O, Pantogny J, Maziere J-C, Bourdillon C (2005) Langmuir 21:4661–4668
Wolf LK, Gao Y, Georgiadis RM (2007) J Am Chem Soc 129:10503–10511
Tokuhisa H, Liu J, Omori K, Kanesato M, Hiratani K, Baker LA (2009) Langmuir 25:1633–1637
Kim K, Yang H, Jon S, Kim E, Kwak J (2004) J Am Chem Soc 126:15368–15369
Ryan D, Parviz BA, Linder V, Semetey V, Sia SK, Su J, Mrksich M, Whitesides GM (2004) Langmuir 20:9080–9088
Buxboim A, Bar-Dagan M, Frydan V, Zbaida D, Morpurgo M, Bar-Ziv R (2007) Small 3:500–510
Jonkheijm P, Weinrich D, Köhn M, Engelkamp HE, Christianen PCM, Kuhlmann J, Maan JC, Nüsse D, Schroeder H, Wacker R, Breinbauer R, Niemeyer CM, Waldmann H (2008) Angew Chem Int Ed 47:4421–4424
Ballav N, Schlip S, Zharnikov M (2008) Angew Chem Int Ed 47:1421–1424
Winkler T, Ballav N, Thomas H, Zharnikov M, Terfort A (2008) Angew Chem Int Ed 47:7238–7241
Ballav N, Terfort A, Zharnikov M (2009) J Phys Chem C 113:3697–3706
Ballav N, Terfort A, Zharnikov M (2009) Langmuir 25:9189–9196
Jung LS, Nelson KE, Stayton PS, Campbell CT (2000) Langmuir 16:9421–9432
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Frasconi, M., Mazzei, F. & Ferri, T. Protein immobilization at gold–thiol surfaces and potential for biosensing. Anal Bioanal Chem 398, 1545–1564 (2010). https://doi.org/10.1007/s00216-010-3708-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-3708-6