
Abstract
This paper describes a method for determination of 27 mycotoxins and other secondary metabolites in maize silage. The method focuses on analytes which are known to be produced by common maize and maize-silage contaminants. A simple pH-buffered sample extraction was developed on the basis of a very fast and simple method for analysis of multiple pesticide residues in food known as QuEChERS. The buffering effectively ensured a stable pH in samples of both well-ensiled maize (pH < 4) and of hot spots with fungal infection (pH > 7). No further clean-up was performed before analysis using liquid chromatography–tandem mass spectrometry. The method was successfully validated for determination of eight analytes qualitatively and 19 quantitatively. Matrix-matched calibration standards were used giving recoveries ranging from 37% to 201% with the majority between 60% and 115%. Repeatability (5–27% RSDr) and intra-laboratory reproducibility (7–35% RSDIR) was determined. The limit of detection (LOD) for the quantitatively validated analytes ranged from 1 to 739 µg kg−1. Validation results for citrinin, fumonisin B1 and fumonisin B2 were unsatisfying. The method was applied to 20 selected silage samples and alternariol monomethyl ether, andrastin A, alternariol, citreoisocoumarin, deoxynivalenol, enniatin B, fumigaclavine A, gliotoxin, marcfortine A and B, mycophenolic acid, nivalenol, roquefortine A and C and zearalenone were detected.

Maize silage fed to cows can be contaminated with mycotoxins from pre- and post-harvest fungi. Several metabolites were detected by LC-MS/MS; including zearalenone and mycophenolic acid from Fusarium (red) and Penicillium (green) infections, respectively.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Maize silage is contaminated with a wide variety of pre- and post-harvest fungi, which may lead to undesired production of mycotoxins and other secondary metabolites [1]. The intake of mycotoxins may affect animal health and productivity [2]. Transfer of various mycotoxins from feed to milk and meat is also of concern [3, 4]. In modern dairy farming systems, dairy cows are consuming up to 40 kg/day, with maize silage constituting 50–75% of the diet [5].
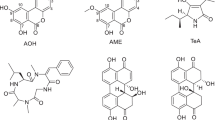
Pre-harvest fungal contaminants of maize plants under Danish conditions include mainly Fusarium and Alternaria species [6] whereas post-harvest contaminants of maize silage include Penicillium roqueforti, Penicillium paneum, Byssochlamys nivea and Aspergillus fumigatus [7]. These species are in culture capable of producing a range of chemically very diverse compounds (Fig. 1), ranging from: (1) small polar but neutral compounds like patulin; (2) acidic compounds like mycophenolic acid and hydroxyl-benzoic acids; (3) basic compounds like roquefortines and marcfortines; and (4) large apolar compounds like penitrems and enniatins [1].

Structural differences and various functional groups of the compounds tested and their names and abbreviations
Due to the chemical differences of the fungal metabolites, multi-mycotoxin methods with no sample clean-up are needed. Such methods have been reviewed by Zöllner and Mayer-Helm [8] and Krska et al. [9]. These multi-methods mainly include regulated toxins in food and feed, e.g. aflatoxins B1, B2, G1, G2, M1, ochratoxin A, patulin, deoxynivalenol, zearalenone, fumonisins B1 and B2, T-2 and HT-2 toxin [10]. Most of the multi-methods use LC-MS/MS on triple quadrupole systems, although LC-TOF-MS and LC-Orbitrap based methods are also looking promising [11].
Altogether, few multi-mycotoxin methods have been fully validated in silage [12, 13]. Ideally, a method for the screening of maize silage samples should include the whole secondary metabolic potential of the pre- and post-harvest contaminants with an emphasis on the mycotoxins. Driehuis et al. [12] measured 20 analytes of which five are post-harvest compounds (ochratoxin A, mycophenolic acid, penicillic acid, roquefortine C, sterigmatocystin), but none of these are associated with A. fumigatus or P. paneum.
Garon et al. [13] detected seven mycotoxins of which only citrinin and gliotoxin originate from common post-harvest contaminants of silage. The studies by Mansfield et al. [14] and O’Brien et al. [15] both use LC-TOF-MS, and focus on the metabolites from the penicillia, but none were validated adequately. Mansfield et al. did not include any qualifier ions nor used the high-resolution capability of the instrument, and the study of O’Brien et al. was mainly focusing on novel compounds like marcfortine A and andrastin A.
Maize silage is a complex matrix as the whole maize plant is fermented. It contains, e.g., chlorophylls and carotenoids from the leafy parts of the plant, starch, glucans from the cob and organic acids from the ensiling, and is thus much more complex than products based on the maize kernels. Since pH may vary from 3.6 in well-ensiled maize to 7–9 in fungal hotspots [16], pH must be controlled by buffers when extracting and analysing silage samples. pH changes the polarity of compounds with ionisable groups [17], thus affecting the extraction efficiency.
Trace analysis of pesticide residues in fruit, vegetables and cereals is in many ways comparable to mycotoxin analysis. An increasingly popular multi-method for pesticide detection in various matrixes is the Quick, Easy, Cheap, Effective, Rugged and Safe method, known as QuEChERS [18–20]. Briefly, the method uses acetonitrile for extraction of the analytes followed by the addition of high concentrations of MgSO4 and NaCl. The salts induce a phase separation between ACN and water, keeping extremely polar contaminants in the water. Buffering can be applied to overcome pH effects of the matrix on the extraction efficiency of chargeable compounds [21].
The aim of the present study is to adapt, apply and evaluate the QuEChERS method to the extraction of multiple mycotoxins in maize silage samples. To our knowledge, it is the first publication describing the use of QuEChERS in mycotoxin analysis. A LC-MS/MS method was developed and validated for the detection of mycotoxins in the silage extracts. The method targets chemically very different metabolites from pre-harvest (Fusarium culmorum, Fusarium graminearum, Fusarium avenaceum and Alternaria tenuissima) and post-harvest (A. fumigatus, Monascus ruber, P. roqueforti, P. paneum, B. nivea) fungal contaminants of silage. The structures, names and abbreviation of the tested compounds are shown in Fig. 1. The method has been applied to 20 field samples of naturally contaminated maize silage.
Experimental
Chemicals and reagents
Acetonitrile (ACN) and methanol (MeOH) were HPLC-grade (Rathburn, Walkerburn, Scotland, UK). NaOH, CH3COOH, HCOOH, NH4OH, HCOONH4, CH3COONa, MgSO4 and CH3COONH4 were all of analytical reagent grade. Water was ultra-purified using a Millipore system (Molsheim, France).
Standards were purchased from commercial suppliers; FUT C, ENN B from Alexis Biochemicals (Farmingdale, NY, USA), AOH, AME, CIT, NIV, DON, GLI, MPA, CPA, OTA, PAT, ROQ C, T-2, TEA, ZEA, MEV, PEN A and STE all from Sigma-Aldrich (St. Louis, MO, USA). FB1 and FB2 were acquired from Romer Labs (Tulln, Austria). Quantitative standards of AND A and FUT A and B as well as qualitative standards of CICO, FUC A and B, MAC A and B, and PR were available for LC-MS/MS optimisation from earlier studies [22].
For the spiking experiments, a pooled fungal extract was prepared. Agar cultures of A. tenuissima, P. roqueforti, P. paneum and A. fumigatus were extracted according to Smedsgaard [23] with a few modifications. ALS was only available from fungal extract of A. tenuissima, but was confirmed by LC-TOF-MS and UV characteristics [24].
All mycotoxin stock solutions (25–5,485 µg·mL−1) were prepared in ACN and kept at −18 °C unless otherwise recommended by the manufacturer.
Sample preparation
Silage samples were frozen with liquid N2 and homogenised in a domestic blender. Extraction was performed by a modified version of a method for multiple pesticide residues in food known as QuEChERS [21]: In a 50-mL polypropylene tube, 10.0 g sample (fresh weight) was extracted with a buffered mixture of 10 ml 1% acetic acid in ACN, 5 ml water and 1.67 g sodium acetate tri-hydrate by shaking for 1–2 min. Then, 4.0 g anhydrous MgSO4 was added and the tube was shaken (1 min) to obtain phase separation. After a 10-min centrifugation (4,500×g), the upper ACN phase was collected. Before LC-MS/MS analysis, the samples were filtered through a 0.45 μm PFTE filter in Mini-UniPrep HPLC vial (Whatman International, Maidstone, Kent, UK).
Robustness of buffering
The effectiveness of the buffering incorporated in the method was tested with 12 portions of a silage sample naturally contaminated with P. roqueforti/P. paneum which were also spiked with pure standards at medium level according to the validation plan. The pH of six portions of silage was adjusted to pH>10 by adding 5 ml of 0.55 M NaOH in the first extraction step instead of 5 ml of water. Triplicate samples at both the natural silage pH of 4.2 and pH > 10 were subjected to either a traditional extraction with a 4:1 (v/v) unbuffered mixture of ACN and water or the present method. Extracts were analysed by LC-MS/MS and pH in the surplus extracts was measured after dilution 1:4 (v/v) with water. The effect of pH on analyte response with each extraction method was evaluated with the PROC GLM procedure in SAS 9.1 (SAS Institute Inc., Cary, NC).
LC-MS/MS method
LC separation of 1 μL injected sample was performed on an Agilent 1100 series HPLC system (Agilent Technologies, Palo Alto, CA, USA) with a Gemini C6-Phenyl, (3 μm, 2.0 × 100 mm) column equipped with a Gemini Security guard cartridge (Phenomenex, Torrance, CA, USA). Samples were analysed in two separate runs, one in positive electrospray ionisation (ESI) mode and one in negative ESI mode. Water-based HPLC eluents were prepared daily. The mobile phases were (A) ammonium formiate 0.4 mM, 0.2% formic acid in water (pH 2.5) and (B) 100% ACN for data recorded in ESI+. In ESI−, they were (A) 0.02% formic acid in water and (B) 100% ACN. The gradient conditions were identical. During data collection, a flow rate of 0.3 ml/min of was used: from 0 to 4 min 10% B was kept constant, then going to 100% B from 4 to 22 min. The LC-system and column was cleaned after each sample, by injecting three different blanks: (1) 20 μL 5% formic acid in ACN with 100% B at 0.5 ml/min for 8 min; (2) then 20 μL methanol and gradually changing to 10% B and 0.3 ml/min in 5 min; (3) and finally, 20 μL water maintained at 10% B and 0.3 ml/min for 7 min. This gave a total runtime of 44 min per sample. To protect the MS interface, a valve integrated with the MS instrument was used to direct the eluent into the MS instrument only from 1 to 22 min of the gradient. The auto sampler and column temperature was 25 °C.
A Quattro Ultima triple quadrupole MS without the high-collision hexapole (Waters, Manchester, UK) with Masslynx v. 4.1 software was used for data collection and processing. The MS was tuned to symmetrical peak shapes with a peak width of 0.5 mass unit at half peak height. The capillary voltage was 3.0 kV. The source and desolvation temperatures were 120 °C and 400 °C, respectively. The cone gas flow was 80 l h−1 and the desolvation gas flow was 530 l h−1. Argon was used as collision gas at ∼2.5 × 10−3 mbar and the electron multiplier voltage applied was 650 V. Fragment ion spectra were recorded from 15–50 V in both polarities and promising selective fragment ions tested and optimised along with the cone voltage in the multiple-reaction monitoring (MRM) mode. Analyte specific detection parameters are listed in Table 1. Inter channel delay was 0.02 s and the dwell times were optimised for the individual transitions and in the range 100–500 ms.
The response was calculated as the chromatographic peak area for all compounds, except for PAT where height were used. A linear calibration curve was obtained by plotting the response of the analyte against the concentration (c) weighted 1/c. The spiking levels were toxin-specific and were intended to be near the expected detection limit.
Validation set-up
A total of three series were performed by two different technicians on three separate days. Each series included three blind samples, three replicates of samples spiked quantitatively at low, medium and high level and three replicates of samples spiked with a fixed volume of fungal mixture. The spiking levels for each analyte are described in Table 2. Three samples from one well-ensiled Danish maize silage stack with low toxin content were pooled and used as blank and for spiking. The blank silage had traces of 5 ppb ENN B, which was determined using standard addition. The matrix-matched calibration curve of the quantitative standards included six concentration levels and a blank matrix extract. The six levels were obtained by serial dilution with at dilution factor of 128 from the highest to the lowest level. One matrix-matched fungal standard equal to the fungal spike level was also included. Standards were analysed twice; in the beginning and at the end of each sequence. From the results obtained, the relative standard deviation under repeatability conditions (RSDr), intra-laboratory reproducibility conditions (RSDIR), and recovery (Rec.) was calculated for each compound according to ISO guidelines [25]. RSDr and RSDIR represent the variation between repeated extractions and analysis within days and between days, respectively. We accepted results from spiking levels when the RSDIR was up to 35%. For compounds quantitatively available, the limit of detection (LOD) was determined as three times the standard deviation at intra-laboratory conditions (SDIR) divided by the recovery, both based on results from the lowest accepted spike level.
Signal suppression and enhancement (SSE) due to matrix compounds was evaluated as the slope of a standard curve in pure ACN divided by the slope of a standard curve in blind matrix extract (α ACN/α matrix).
Sample analysis
Samples were extracted and analysed according to the method described above. A matrix-matched calibration curve was produced and included in each sample series. The blank silage extract for the calibration curve was produced together with the sample extracts on the basis of aliquots of the blank silage used for validation. The standards were distributed randomly over the entire sequence and used for quantification in the present series.
Sample data was processed by Quanlynx and subjected to (a) visual inspection of un-smoothed chromatogrammes for low concentration samples to determine whether peaks were above a signal to noise of 3:1 (b) visual inspection of the automatic integrations, with manual modifications to consistent peak width if necessary.
Results and discussion
Extraction and clean-up
The application of the adapted QuEChERS method to mycotoxin extraction was successful. Comparing to the extraction methods employed in [12, 26], these methods employ ACN (or MeOH) with 10–20% (v/v) water, while the present method has 33% (v/v) mix of water and ACN. This should allow for better extraction of the more polar analytes. With the induction of phase separation the extraction of less polar compounds is facilitated. According to [18], the ACN phase holds approximately 8% of water. The high concentration of salt in the water phase forces the polar analytes into the less polar ACN. In the case of varying water content in silage samples, the phase separation should also result in a more stable polarity in the extract. This is of relevance as fungal hot spots in silage are much wetter than non-infected silage due to the microbial activity.
The buffering incorporated in the method was very effective. In spiked silage samples adjusted to pH > 10 and subsequently subjected to our buffered modified QuEChERS extraction, the pH of the ACN phase (diluted 1:4 v/v with water) was 4.3. In the same silage, at its natural pH of 4.2, the pH of the ACN phase was 3.7. When the same silage samples were subjected to traditional extraction with an unbuffered mixture of ACN and water the corresponding pH values of the extract were 10.4 and 4.4. The substantial difference in pH had effects on the analysis with both extraction methods. For AOH, CPA, FUC A, FUC C, PEN A and ZEA a significant difference in LC-MS/MS response was observed between the low- and high-pH samples extracted with the unbuffered mixture of ACN and water, with P values of 0.001, 0.029, 0.003, 0.047, 0.002 and 0.001, respectively. For these analytes, no significant difference in response was observed with the buffered QuEChERS method. The response of the analytes AND A, CICO, OTA and ROQ C differed significantly between the two pH values for both extraction methods (P values ranging from 0.047 to <0.001), while the responses for FUT A and STE only differed significantly between pH values when extracted according to the QuEChERS method (P = 0.047 and 0.027, respectively).
It is possible that the extraction of field samples can be improved by performing a longer initial extraction without any salts. However, Lehotay et al. [21] did not experience any negative effect of combining the extraction and partitioning steps into one procedure. Both pesticides and fungal metabolites may be present inside and outside the plant depending on application methods and infection biology, respectively. Therefore, some analytes may be less accessible to extraction than others. Hence, the very short extraction time (<2 min) may pose a problem which is not addressed in validation with spiked samples. Optimisation of extraction time should therefore be done with naturally infected samples. However, several metabolites were detected when the present method was applied to naturally contaminated samples (Table 3).
The amount of sodium acetate or sodium chloride in the QuEChERS method is known to influence the extraction of both analytes and matrix compounds [18, 21]. Fine-tuning of this concentration has not been done but might improve the balance between analytes and interferences in the extract.
The dispersive solid-phase extraction (SPE) with primary–secondary amine (PSA) employed in the QuEChERS methods for pesticides [18, 21], was not used for mycotoxins. PSA binds organic acids, which in our case would be MPA, OTA and CIT as well as TEA and CPA, which also have acidic properties. It was therefore chosen not to employ this clean-up procedure.
In the initial steps of the multi-method development, SPE was tested for clean-up of silage extract. Both C18, polymeric (Strata X) and mixed mode columns (Strata X-C, Oasis MAX) were tested without satisfactory results. In the application of reverse-phase SPE (C18) only few matrix components could be removed to fit all compounds. When also taking advantage of the functional groups on the molecules several analytes were not retained very well and the procedure was very time-consuming. Even in combined extracts from the SPE clean-up, large quantities of matrix were still present. In some cases, regulation of pH in the extracts in order to optimise SPE retention also led to phase separation of the extract, which interferes with the SPE separation. SPE clean-up did therefore not constitute an improvement.
LC-MS-MS method
The compounds were MS-tuned (Table 1) in their most sensitive ESI mode (+/−). However, for MPA, OTA, ROQ C data have been collected in both modes. The ESI− was preferred for these compounds due to better recoveries and lower RSDs and LODs. The better sensitivity of these analytes in ESI− than in ESI+ was unexpected, especially since OTA and ROQ C in solvent have been found to be much greater than tenfold more sensitive in ESI+ than in ESI− on a Micromass LCT and an almost identical Quattro Ultima triple quadrupole MS (in this case equipped with a high-collision hexapole).
From LC-MS/MS runs of single standards, it was concluded that the compounds in the method did not interfere with each other in the measuring range. At AME concentrations much higher than the validated measuring range, some interference with ZEA was observed. This was possible due to co-elution and because AME formed a small amount of the adduct [M + HCOO]− having the same mass (m/z 317) as the ZEA [M-H]− precursor. The ion ratio (quantifier/qualifier) of the interfering daughter ions from AME (2.3) was however different from the ratio of ZEA (0.8). Unique ion-transitions were preferred for the MS/MS method. Daughter ions resulting from water or adduct loss were avoided when possible.
The evaluation of SSE due to matrix compounds showed a large variation in the matrix effect between analytes. Results for the quantitatively validated analytes are presented in Table 4. The signal for MEV is highly enhanced by the silage extract while AOH and ROQ C signals are suppressed. The use of matrix-matched calibration standards compensated for the matrix effects. For TEA, the sensitivity in pure ACN standards was insufficient for evaluation of SSE. MPA and OTA also showed a highly concentration-dependent matrix effect.
The post-run cleaning procedure with injections of formic acid in ACN, MeOH and water was necessary to prevent matrix build-up on the column. Without the procedure, unstable RTs and rapid decreases in sensitivity were observed after just eight injections of silage extract. To continually monitor a potential matrix accumulation on the chromatographic column, a blank ACN sample was included after nine matrix samples injected. These steps gave reliable and stable MS/MS signals throughout a sequence. To achieve lower RSD in the MS analysis, shorter sequences (<24 h) and correction by internal standards for the individual compounds should be applied.
To ensure proper formation of ammonium adducts (T-2 and ENN B) and to obtain better chromatography of the pH-dependent compounds (e.g. ROQ A, CPA, CIT) eluent A used in ESI+ was added ammonia and formic acid. Eluent A for ESI− had only low formic acid content and application of ammonium formiate buffer resulted in a significant signal suppression of the early eluting compounds. This was not accepted as NIV, DON, PAT and GLI already were expected to have high detection limits. It was therefore decided to analyse samples in two separate runs, even though the instrument can switch between the two modes. Switching between positive and negative ionisation requires extra time for data collection. This can, in practice, cause troubles for quantification due to few data points across the peaks and short dwell times when several compounds co-elute [26]. By the use of separate retention time windows for the two ionisation modes, Berthiller et al. [27] simultaneously determined several mycotoxins in a single run. However, because of co-elution and sensitivity drop using common eluent A, this approach was not applicable to our method and instead we accepted a longer instrument time (2 × 44 min) for every sample.
Method performance
For compounds with little matrix interference, the calculation of LOD on the basis of SDIR at lowest accepted spike level gave a higher and probably more realistic detection limit than if based on noise in blind maize silage. The maximum content in feed of 2,000 µg kg−1 ZEA, 8,000 µg kg−1 DON and 250 µg kg−1 OTA recommended by the European Commission [28] can easily can be determined with the current LODs, also when accounting for the differences in dry matter content in the EC recommendation and the present method.
Previous examinations of mycotoxins in maize silages have detected ZEA and DON [12], ENN B [29] and PAT, MPA, CPA and ROQ C [14]. The average toxin concentrations (range in parentheses) in these studies were: ZEA 174 µg kg−1 (25–943), DON 854 µg kg−1 (250–3,142), ENN B 73 µg kg−1 (24–218), PAT 80 µg kg−1 (10–1,210), MPA 160 µg kg−1 (20–1,300), CPA 120 µg kg−1 (20–1,430) and ROC C 380 µg kg−1 (10–5,710). LOD of the current method (corrected for a dry matter content of 35% where appropriate) for ZEA, ENN B and MPA were at level with or below the reported concentration levels. However, for DON, PAT, CPA and ROQ C monitoring of silage with our method is known only to identify part of the samples with the toxins present, as the current LODs are higher than some the of reported contents. Still, it is relevant to measure the frequency of these analytes in more contaminated samples.
Precision and recovery
Compounds with accepted validation results are included in Table 2. Some average recoveries were outside the preferred range of 70% to 110% [30] and still accepted in this multi-method. Optimal extraction and detection of all analytes are not always achievable when several compounds are targeted [26, 27]. We accepted a RSDIR up to 35%, though <22–23% is normally preferred in the 100 ppb range [30–32]. A high RSDIR results in a large uncertainty range, when reporting results. In the application of the method, the detection of the compounds is just as important as a very narrow concentration range in reporting the result. The method was developed for research purposes, not for official food and feed control.
The European Commission [33] has specific criteria for analytical methods applied in foodstuffs (not feed) for a few toxins. Enniatin B, OTA and zearalenone comply with all the specific criteria. The recovery of T-2 (55%) is a little lower than the demanded range (60–130%) and the RSDr of PAT (17%) is slightly higher than the 15% accepted. The average recoveries of 6% FB1 and 13% FB2 were far below the accepted 70–110% range [33]. Methods focusing only on these water-soluble toxins by using immunoaffinity columns have approximately 100% recoveries [34]. However, acceptable recoveries of FB1 and FB2 were also achievable when acidified solvent was used to extract multiple toxins from breadcrumb matrix [26]. The validation of CIT was unsatisfying due to LC-MS/MS instrument day-to-day variations. Results from day 1 showed that CIT is extracted with the QuEChERS method, as spiking at 200 µg kg−1gave 65% mean recovery with 23% RSDr (n = 3). However, on days 2 and 3, a decrease in sensitivity for CIT during the sequence gave unacceptable standard curves and recoveries. Applying LC-MS Garon et al. [13] were able to validate CIT in SPE cleaned-up silage extracts using a HPLC gradient with ACN and acidified water (0.5% acetic acid, pH 3) as mobile phases. This indicates that removal of more matrix components or the use of more acidic eluents than in the present method could be important for proper detection of CIT.
The recoveries were within the same range for low, medium, high and fungal spike and for days 1, 2 and 3 for most compounds. Concentration dependence was only seen for TEA as the recoveries were 52%, 30% and 28% for fungal, 200 µg kg−1 and 404 µg kg−1 respectively. The fungal spike of TEA was much higher than the other two levels near the LOD. On the second validation day the analytes AND A, CPA, FUC C and ROQ C showed unacceptably high day-to-day variations in recovery. These data were omitted (see footnote to Table 2) and instead an additional fungal spike was carried out. High reproducibility was observed for; ROQ A (81%, 138%, 90%), PR (55%, 44%, 69%) and CPA (50%, omitted, 76%) here expressed as the day-to-day variation in mean recovery. It is recommended always to include spiked control samples to evaluate the recovery of the analysis series.
The LC-MS/MS method detects FUC B and FUT C, but they were not present in sufficient amount in the fungal extract to be validated. Their extraction is expected to be like the compounds with structural similarities (FUC A and FUT A, respectively). Similarities in validation data are observed for the isomers ROQ A and FUC A and for the closely related AOH and AME (–OH/–CH3 group).
Identification criteria
Ideally, identification of a compound should fulfil certain criteria: a retention time (RT) tolerance of 3%, a signal to noise ratio of at least three and similar relative abundances of the diagnostic ions as for spiked samples. At spike levels close to LOD, some ion ratios varied more than recommended in [30] (see footnote to Table 2).
Quantitative ions from mycotoxins spiked at the lowest accepted level (Fig. 2) and silage spiked with the fungal mix (Fig. 3) have been compared to the signal of blank silage. The blank silage was selected among available maize silage samples to have a low natural content of mycotoxins. The smooth chromatograms of the quantitative ions show a low noise for most compounds. Figures 2 and 3 also visualise the broad peak of TEA and FUM A and matrix interference on the quantitative ion of PAT. To overcome a large closely eluting matrix interference for the quantitative ion of PAT with m/z 109 (Fig. 2), the peak height was used as response variable instead of peak area. No matrix interference was observed for the less sensitive product ion (m/z 81). Using height instead of area enabled automatic integration and ensured acceptance of ion ratio (±20% of standards) for PAT in all spiked samples. NIV and DON were identified by their RTs only. The ion ratios in the matrix-matched standards could not be verified for the major part of the spiked NIV and DON samples due to the qualifier's low sensitivity and interference of matrix.

The relative abundances of the MRM chromatogram traces for the quantitative ions of the mycotoxins in blank maize silage and spiked at the lowest accepted level with quantitative standards

The relative abundances of the MRM chromatogram traces for the quantitative ions of the mycotoxins in blank maize silage and silage spiked with a mixture of fungal extracts
Field samples
The method was applied to 20 naturally contaminated samples of maize silage collected at Danish dairy farms (Table 3). Ten samples were visibly un-mouldy samples extracted with a silage drill while ten were hot spots with visible fungal growth collected from the cutting face of the silages. The mycobiota of the selected samples was determined previously [7; unpublished data] and the hot spots selected to represent a range of the most common post-harvest contaminants of silage: P. roqueforti, P. paneum, A. fumigatus, B. nivea and M. ruber [1]. Reported are compounds which were above the LOD in either the unspoiled silage or in the fungal hot spots and met the identification criteria. The P. roqueforti/P. paneum metabolites AND A and CICO were very common in both hot spots and visibly uninfected silages. Likewise, ROQ A was detected in both types of samples, while MPA and ROQ C were most common in hot spots with maximum concentrations (±95% confidence interval) of 1,646 (±460) µg kg−1 and 37 (±18) mg kg−1, respectively. The high concentrations of MPA, ROQ C and AND A in some hot spots are consistent with observation in grass silage by O’Brien et al. [15]. ROQ C was generally present in samples infected with P. roqueforti in accordance with Auerbach et al. [35]. AND A could be a good marker for Penicillum spoilage during storage because of the low detection limit and its detection in the majority of the unspoiled silages. The maximum concentration of MPA was in a hot-spot infected by B. nivea. This fungus is known to produce MPA [36] and this result shows that it is also capable of producing it in silage. The two hot spots with growth of A. fumigatus contained GLI in concentrations up to 906 (±245) µg kg−1. FUC A, another known A. fumigatus metabolite, was also detected in one of these samples. ZEA, NIV, DON and ENN B are toxins from Fusarium species infecting maize pre-harvest [6]. They have also been detected with the current method. The concentrations were near the limit of detection and much below the maximum contents in feed recommended by the European Commission [28].
Conclusion
A new method for detection of 27 fungal secondary metabolites in maize silage was developed and successfully validated. Nineteen of the analytes can be detected quantitatively and eight qualitatively with recoveries from 37 to 201%, LODs from 1 to 739 μg kg−1 and reproducibilities from 7 to 35%. The pH-buffered extraction method ensured the same extraction conditions for fungal hot spots (pH > 7) and normal silage (pH ∼ 4). Applied to 20 Danish maize silage samples, the following mycotoxins and other fungal secondary metabolites were detected: AME, AND A, AOH, CICO, DON, ENN B, FUC A, GLI, MAC A, MAC B, MPA, NIV, ROQ A, ROQ C and ZEA, representing metabolites from common fungal pre- and post-harvest contaminants of maize silage. With this application of the QuEChERS method to mycotoxin analysis, it may in the future be possible to combine mycotoxin and pesticide analysis.
References
Storm IMLD, Sørensen JL, Rasmussen RR, Nielsen KF, Thrane U (2008) Mycotoxins in silage. Stewart Posthar Rev 4. doi:10.2212/spr.2008.6.4
Fink-Gremmels J (2008) The role of mycotoxins in the health and performance of dairy cows. Vet J 176:84–92
Fink-Gremmels J (2008) Mycotoxins in cattle feeds and carry-over to dairy milk: a review. Food Addit Contam 25:172–180
Miller JD (2008) Mycotoxins in small grains and maize: old problems, new challenges. Food Addit Contam 25:219–230
Driehuis F, Spanjer MC, Scholten JM, Giffel MCT (2008) Occurrence of mycotoxins in feedstuffs of dairy cows and estimation of total dietary intakes. J Dairy Sci 91:4261–4271
Sørensen JL (2009) Preharvest fungi and their mycotoxins in maize. PhD thesis. Technical University of Denmark, Kgs. Lyngby
Storm IMLD (2009) Post-harvest fungal spoilage of maize silage - Species, growth conditions and mycotoxin detection. PhD thesis. Technical University of Denmark, Kgs. Lyngby, Denmark
Zöllner P, Mayer-Helm B (2006) Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography-atmospheric pressure ionisation mass spectrometry. J Chrom A 1136:123–169
Krska R, Schubert-Ullrich P, Molinelli A, Sulyok M, Macdonald S, Crews C (2008) Mycotoxin analysis: an update. Food Addit Contam 25:152–163
Commission EC (2006) Commission regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off J Eur Union L364:5–24
Herebian D, Zuhlke S, Lamshoft M, Spiteller M (2009) Multi-mycotoxin analysis in complex biological matrices using LC-ESI/MS: experimental study using triple stage quadrupole and LTQ-Orbitrap. J Sepa Sci 32:939–948
Driehuis F, Spanjer MC, Scholten JM, Giffel MCT (2008) Occurrence of mycotoxins in maize, grass and wheat silage for dairy cattle in the Netherlands. Food Addit Contam B 1:41–50
Garon D, Richard E, Sage L, Bouchart V, Pottier D, Lebailly P (2006) Mycoflora and multimycotoxin detection in corn silage: experimental study. J Agr Food Chem 54:3479–3484
Mansfield MA, Jones AD, Kuldau GA (2008) Contamination of fresh and ensiled maize by multiple penicillium mycotoxins. Phytopathology 98:330–336
O'Brien M, Nielsen KF, O'Kiely P, Forristal PD, Fuller HT, Frisvad JC (2006) Mycotoxins and other secondary metabolites produced in vitro by Penicillium paneum Frisvad and Penicillium roqueforti Thom isolated from baled grass silage in Ireland. J Agr Food Chem 54:9268–9276
Müller HM, Amend R (1997) Formation and disappearance of mycophenolic acid, patulin, penicillic acid and PR toxin in maize silage inoculated with Penicillium roqueforti. Arch Anim Nutri 50:213–225
Kah M, Brown CD (2008) Log D: lipophilicity for ionisable compounds. Chemosphere 72:1401–1408
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86:412–431
Lehotay SJ, de Kok A, Hiemstra M, van Bodegraven P (2005) Validation of a fast and easy method for the determination of residues from 229 pesticides in fruits and vegetables using gas and liquid chromatography and mass spectrometric detection. J AOAC Int 88:595–614
Lehotay SJ (2007) Determination of pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulfate: collaborative study. J AOAC Int 90:485–520
Lehotay SJ, Mastovska K, Lightfield AR (2005) Use of buffering and other means to improve results of problematic pesticides in a fast and easy method for residue analysis of fruits and vegetables. J AOAC Int 88:615–629
Nielsen KF, Smedsgaard J (2003) Fungal metabolite screening: database of 474 mycotoxins and fungal metabolites for dereplication by standardised liquid chromatography-UV-mass spectrometry methodology. J Chrom A 1002:111–136
Smedsgaard J (1997) Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. J Chrom A 760:264–270
Hellwig V, Grothe T, Mayer-Bartschmid A, Endermann R, Geschke FU, Henkel T, Stadler M (2002) Altersetin, a new antibiotic from cultures of endophytic Alternaria spp. Taxonomy, fermentation, isolation, structure elucidation and biological activities. J Antibio 55:881–892
ISO (1994) 5725-2: Accuracy (truness and precision) of measurement methods and results. Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method
Sulyok M, Krska R, Schuhmacher R (2007) A liquid chromatography/tandem mass spectrometric multi-mycotoxin method for the quantification of 87 analytes and its application to semi-quantitative screening of moldy food samples. Anal Bioanal Chem 389:1505–1523
Berthiller F, Schuhmacher R, Buttinger G, Krska R (2005) Rapid simultaneous determination of major type A- and B-trichothecenes as well as zearalenone in maize by high performance liquid chromatography-tandem mass spectrometry. J Chrom A 1062:209–216
Commission EC (2006) Commission recommendation of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 and HT-2 and fumonisins in products intended for animal feeding. Off J Eur Union L229:7–9
Sørensen JL, Nielsen KF, Rasmussen PH, Thrane U (2008) Development of a LC-MS/MS method for the analysis of enniatins and beauvericin in whole fresh and ensiled maize. J Agr Food Chem 56:10439–10443
Commission EC (2002) Commission decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Eur Commun L221:8–36
Thompson M (2000) Recent trends in inter-laboratory precision at ppb and sub-ppb concentrations in relation to fitness for purpose criteria in proficiency testing. The Analyst 125:385–386
Horwitz W (1982) Evaluation of analytical methods used for regulation. J Assoc Off Anal Chem 65:525–530
Commission EC (2006) Commission regulation (EC) No 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off J Eur Union L70:12
Kim EK, Maragos CM, Kendra DF (2004) Liquid chromatographic determination of fumonisins B1, B2, and B3 in corn silage. J Agr Food Chem 52:196–200
Auerbach H, Oldenburg E, Weissbach F (1998) Incidence of Penicillium roqueforti and roquefortine C in silages. J Sci Food Agric 76:565–572
Puel O, Tadrist S, Galtier P, Oswald IP, Delaforge M (2005) Byssochlamys nivea as a source of mycophenolic acid. Appl Environ Microbiol 71:550–553
Acknowledgements
Funding for this study was provided by The Directorate for Food, Fisheries, and Agri Business (Copenhagen, Denmark (#FFS05)), Danish Cattle Federation, the Research School FOOD and the Technical University of Denmark.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rasmussen, R.R., Storm, I.M.L.D., Rasmussen, P.H. et al. Multi-mycotoxin analysis of maize silage by LC-MS/MS. Anal Bioanal Chem 397, 765–776 (2010). https://doi.org/10.1007/s00216-010-3545-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-3545-7