
Abstract
The cellular behavior of ginsenosides on cancer cells has not been measured directly despite their potent anticancer activities and biological actions. A liquid chromatography–mass spectrometry (LC-MS) method was developed to measure the selective cellular uptake of ginsenosides in both cell lysates and culture media. Fifteen ginsenosides were separated within 17 min with good peak shapes using a 2-μm sub-particle size C18 column. Quantification was performed by triple-quadrupole MS with electrospray ionization in negative ion mode. The sample preparation containing the solid-phase extraction was linear (correlation coefficient, r 2 > 0.992) for all analytes, while the limit of quantification ranged from 0.5 to 2.0 ng/mL in both matrices. The assay precision (%CV) and accuracy (%bias) at three different concentrations (5, 20, and 100 ng/mL) were 1.4% to 11.6% and 94.9% to 106.4%, respectively. When this method was used to examine the selective cellular uptake of ginsenosides, the relative non-polar and protopanaxadiol class ginsenosides, such as Rg3, Rk1, Rg5, Rh2, compound-K, and protopanaxadiol (PPD), showed cellular uptake in the MCF-7 cells, but the relative polar and protopanaxatriol class of ginsenosides did not accumulate in the cells. The most non-polar ginsenoside PPD, which is an aglycone of the protopanaxadiol type, resulted in the highest uptake rate. These results show that the different anticancer activities are due to the selective uptake of ginsenosides based on their chemical structures. This LC-MS-based method can be used to estimate the biological activity of ginsenosides on cells from their structural diversity.

The structure and the ratios of cellular uptake of ginsenosides evaluated by liquid chromatography–mass spectrometry
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ginsenosides are triterpenoid saponins and the major active compounds of “ginseng,” a herbal medicine derived from the roots and rhizomes of different Panax species (Araliaceae). Approximately 80 ginsenosides have been identified. They are classified mainly by two different aglycones, protopanaxadiol (PPD) and protopanaxatriol (PPT) or are classified by the carbohydrate portions, which are attached to the carbon-3, carbon-6, or carbon-20 in algycone. Ginsenosides have biological effects on various cancer cells, angiogenesis and neurodegenerative disease, and have a variety of biological activities depending on the structural aglycones and carbohydrate portion [1–4]. In particular, PPD-type ginsenosides have higher anticancer activity against cancer cells than the PPT type by triggering apoptotic cell death [5–9].
Although the confocal microscopy with fluorescence and antibodies are used to determine the cellular uptake of chemical compounds [10, 11], the analysis is limited by the need to prepare antibodies and fluorescence labels. In addition, only a single compound can be estimated at a single time. Recently, the qualitative method with the specific antibody on ginsenoside Rg3 was introduced for its uptake on cancer cells [12]. Liquid chromatography–mass spectrometry (LC-MS) was introduced successfully to separate and quantify ginsenosides in biological fluids [13–16]. However, there are no reports of LC-MS-based quantitative analysis of ginsenosides in cell media or cell lysates.
LC-MS techniques have better sensitivity and quantitative reproducibility on the simultaneous analysis of biologically active compounds [17–19]. This paper reports the LC-MS-based quantitative analysis of multiple-ginsenoside cellular uptake on MCF-7 human breast cancer cells. The ginsenosides were selected based on their anticancer activity (Fig. 1) [3, 9]. The aim of this study was to validate a LC-MS quantitative method and evaluate the selective cellular uptake of ginsenosides from white and red ginseng extracts using both cell media and lysates.

Structures of the ginsenosides and selected-ion monitoring (SIM) ions corresponding to m/z [M+CO2H]− of the individual compounds. Glc -d-glucopyranosyl, Ara(p) -l-arabinopyranosyl, Ara(f) -l-arabinofuranosyl, Rha -l-rhamnopyranosyl
Experimental section
Chemicals and reagents
Ginsenosides Rb1, Rb2, Rc, Rd, Rf, Rg1, and Rh1 were purchased from NaturalChem (Yeongju, Korea), and ginsenosides C-K, Re, Rh2, PPD, and PPT were obtained from the EMBO Institute (Seoul, Korea). Ginsenosides Rg3, Rg5, and Rk1 were kindly donated by Professor Yeong Shik Kim (Seoul National University, Seoul, Korea). Digoxin, as an internal standard (IS), was supplied by Sigma (St. Louis, MO, USA). Ginsenoside-enriched fractions were obtained from white (WG) and red ginseng (RG) extracts based on previous method [20]. A Strata-X cartridge (3 mL, 60 mg; Phenomenex, Torrance, CA, USA) was used for solid-phase extraction (SPE). The human breast cancer cells (MCF-7) were obtained from the American Type Culture Collection (Manassas, VA, USA). The MCF-7 cells were cultured in RPMI 1640 media. The antibiotic–antimycotic were purchased from GIBCO (Grand island, NY, USA) and supplemented with 10% fetal bovine serum (JRH, Lenexa, KS, USA). Formic acid (reagent grade) was obtained from Sigma. All organic solvents used were of analytical or HPLC grade and were purchased from Burdick & Jackson (Muskegan, MI, USA). Deionized water was prepared using a Milli-Q purification system (Millipore, Billerica, MA, USA).
Cell culture and uptake studies
The MCF-7 cells were seeded at a density of 1 × 106/mL in 75-cm2 flasks in triplicate for incubation. After 48 h of preincubation, medium was replaced with fresh medium containing each ginsenoside-enriched fractions from WG and RG extracts (at a concentration of 10 and 50 μg/mL), and the cells were further incubated for 0, 6, and 12 h. The MCF-7 cells were then washed twice with D-PBS and scraped from the flasks. Cell pellets and culture media fractions were collected by centrifugation for 10 min at 14,000 rpm and 4 °C. For estimation of cellular uptake, MCF-7 cell pellets obtained from ginsenosides-loaded cells were suspended and stand with lysis buffer (20 mM Tris, pH 7.4; 250 mM NaCl; 2 mM EDTA, pH 8.0; 0.1% NP-40). Ginsenoside stability was also examined by loading 10 μg/mL of the WG and RG extracts into the cell culture medium and incubating them without cells under the same culture conditions. These cell lysates and culture medium were stored at −80 °C until analysis.
LC-MS analysis
LC-MS analysis was performed on a Varian 212-LC system connected to a 320-MS triple–quadrupole mass spectrometer (Varian, Walnut Creek, CA, USA). The ginsenosides were separated through a Hypersil Gold C18 column (50 mm × 2.1 mm i.d., 1.9 μm particle size; ThermoFinnigan, San Jose, CA, USA) at a column temperature and flow rate of 60 °C and 0.35 mL/min, respectively. After injecting 10 μL, the mobile phase consisting of A (0.1% formic acid in 5% acetonitrile) and B (0.1% formic acid in 95% acetonitrile) was controlled as follows: 0 min at 12% B; 0–1.2 min, 12–30% B; 1.2–7 min, 30–40% B; 7–10 min, 40–70% B; 10–11 min, 70–100% B; 11–13 min, 100% B. The gradient was then returned to the initial conditions (12% B) and held for 3.9 min before injecting the next sample.
All ginsenosides and digoxin were analyzed and quantified in selected-ion monitoring (SIM) mode using electrospray ionization in negative mode with formic acid adduct ions [M+CO2H]− (Fig. 1). The mass spectrometer was operated under the following optimized conditions: a needle potential of −3,800 V, shield potential of −425 V, housing temperature of 55 °C, drying gas (nitrogen) temperature of 200 °C, drying pressure of 20 psi, nebulizing pressure (air) of 55 psi, and EM voltage of 1,700 V.
Standard solution and quality-control samples
Stock solutions of all the ginsenoside standards and digoxin were prepared at 10 mg/mL in dimethyl sulfoxide. The working solutions were prepared with methanol at concentrations ranging from 0.1 to 100 μg/mL. All standard solutions were stored at −20 °C until needed. The quality-control (QC) samples were prepared to optimize the SPE procedures and validate the method in two different matrices (cell media and cell lysates). The cell culture media (100 μL) were spiked with ginsenosides (5, 20, and 100 ng/mL) and digoxin (10 ng/mL). The mixtures were diluted with 2 mL PBS (pH 7.4) and were used as the QC samples. The cell lysates (106 cells in the lysis buffer, 100 μL) were also spiked with the ginsenosides (5, 20, and 100 ng/106 cells) and digoxin (10 ng/106 cells). The cell lysates were extracted twice with 2 mL methanol by sonication for 10 min and then centrifuged at 2,500 rpm for 5 min. The supernatant were combined and evaporated under a gentle nitrogen stream at 40 °C. The dried residues were dissolved in 100 μL methanol and diluted with 2 mL PBS before sample preparation with SPE
Optimization of solid-phase extraction
The SPE cartridges were preconditioned with 2 mL of methanol followed by 2 mL of distilled water. Each QC sample was loaded onto a cartridge and washed with 2 mL distilled water. To optimize the additional washing conditions, the individual SPE cartridges were washed with 0%, 20%, 30%, or 40% methanol (2 mL), and the retained ginsenosides were eluted with 100% methanol. For the elution solvent tests, the cartridges were also washed with distilled water, and 20% methanol and analytes were eluted using 60%, 80%, or 100% (4 mL) methanol. The eluates were evaporated to dryness under a gentle nitrogen stream at 40 °C and reconstituted in 100 μL of mobile phase B. The absolute recovery was calculated by comparing the peak area ratios of the extracted samples versus those of their non-extracted counterparts. To maximize extraction efficiencies, two different SPE cartridges, Oasis HLB (3 mL, 60 mg; Waters, Milford, MA, USA) and Strata-X, were evaluated and compared in extraction efficiencies.
Optimization of sample reconstitution
To optimize the reconstitution solvent during LC-MS analysis, one sample set was dissolved in the following concentrations of mobile phase B: 30%, 50%, 70%, 90%, and 100%. Another sample set was dissolved in aqueous methanol (80%, 90%, and 100%). The samples were then injected into the LC-MS system and compared with the absolute recoveries.
Sample preparation
For the cell culture medium samples, 100 μL of the cell media was fortified with 10 μL of digoxin (0.1 μg/mL) and diluted with 2 mL PBS. The SPE cartridges were placed in a device fitted with a peristaltic pump, which was operated at a low flow rate (<1 mL/min). After loading the sample onto a cartridge, the cartridge washed with distilled water (2 mL) and 20% methanol (2 mL), and the ginsenosides were eluted with absolute methanol (2 mL × 2). The eluates were evaporated under a nitrogen stream. The dried extracts were reconstituted with 100 μL of mobile phase B and injected (10 μL) into the LC-MS system. In the cell lysates, the samples (106 cells in lysis buffer, 100 μL) were also spiked with 10 μL of digoxin (0.1 μg/mL). The samples were extracted twice with 2 mL methanol by sonication for 10 min and then centrifuged at 2,500 rpm for 5 min. The supernatant was collected and evaporated to dryness under a gentle nitrogen stream at 40 °C. The dried residues were dissolved in 100 μL methanol, diluted with 2 mL PBS, and used as the cell samples. The diluted samples were prepared using the same procedures employed for the cell media described above.
Method validation
The QC samples containing 15 ginsenosides were quantified based on the individual LC-MS peak area ratios versus that of digoxin as the IS. The calibration samples were made up at 10 different concentrations ranging from 0.1 to 200 ng/mL or ng/106 cells. The limits of detection (LOD) and quantification (LOQ) were determined based on the response at a signal-to-noise (S/N) ratio of 3 and 10, respectively. The precision and accuracy are expressed as the coefficients of variation (%CV) and the percentage relative error (%bias), respectively, at three different concentrations (low, medium, and high) of QC samples based on the individual analyte calibration range. Four replicates were analyzed to determine the within-day reproducibility, whereas the day-to-day reproducibility was measured by analyzing the samples on four different days. The absolute recovery was calculated by comparing the peak area ratios of the extracted samples versus those of their non-extracted counterparts.
Results and discussion
Mass spectrometry of ginsenosides
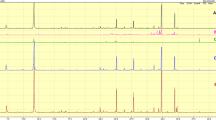
A flow injection of 15 ginsenosides in 50% acetonitrile was used to evaluate the MS responses in both negative and positive ion modes. All ginsenosides were detected with better sensitivity in negative ion mode with [M–H]− ions, and formate adduct ions [M+CO2H]− as the base peak were detected when formic acid was added. Therefore, formic acid was used as an additive to the mobile phase to enhance the ion sensitivity, and the formate adduct ions of all ginsenosides were used in both SIM and selected reaction monitoring (SRM) modes. In SRM analysis, the MS/MS conditions were optimized for the highest S/N ratios of the product ions (see Supplementary Table 1). In the LC-MS/MS chromatograms, the de-formate product ions showed the highest S/N ratios. The other product ions produced mainly by the cleavage of glycosidic bonds, were formed at a high collision energy (CE) level >35 V with a low S/N ratios in all compounds. The MS/MS experiment with the [M–H]− ions without formic acid also showed similar patterns. In addition, the S/N ratios and LOQ levels of each ginsenoside in SRM and SIM analyses were compared with the QC samples of the cell lysates. Although the background noise of SRM mode was lower than SIM mode, the signal intensity was significantly low (Fig. 2). Therefore, SIM mode was chosen to monitor the ginsenosides with both higher S/N ratios and lower LOQ levels of ginsenosides.

The S/N ratios between the SIM and SRM detection modes with a PPD and b ginsenoside Rg1 spiked cell lysates (at 20 ng/106 cells)
Chromatography of ginsenosides
To obtain good detection sensitivity with excellent chromatographic resolution, the described 2-μm sub-particle LC column was introduced, and the gradient separation conditions were optimized [21]. Gradient elution was started at a relatively low percentage of the organic solvent (12%), and increased up to 30% for 1.2 min to wash the other hydrophilic impurities in both the cell lysates and media. The gentle gradient with 30% to 40% for 7 min was used to separate the hydrophobic isomers (Rk1 and Rg5, Rh2 and compound-K) under a 350 μL/min flow rate at a column temperature of 60 °C. The developed LC method enabled the quantification of all ginsenosides within 17 min, which includes the equilibrium time required for the next run (Fig. 3). Although the sample matrices were different from previous reports, this method may be comparable to a three to ten times’ increase in sensitivity [15, 16].

LC-SIM/MS chromatograms of the 15 ginsenosides and digoxin from cell lysates spike with 10 ng/106 cells of PPD, PPT, and Rb1 and 5 ng/106 cells of the others
Sample preparation of cell media and lysates
Efficient sample preparation was required to determine the selective uptake of each ginsenoside by the cells. In SPE experiments, two different SPE cartridges, Oasis HLB and Strata-X, were compared because Oasis HLB resulted in better extraction efficiencies compared with both silica-based C8 and C18 cartridges in ginsenoside analysis [15]. Because Strata-X showed comparable extraction yields with Oasis HLB, Strata-X was chosen in this study (see Electronic supplementary materials Fig. S1). To evaluate the SPE procedures, the absolute recovery of each ginsenoside was measured using the QC samples of cell media spiked at the three different concentrations (5, 20, and 100 ng/mL). After sample loading onto the SPE cartridge and washing with 2 mL of distilled water, the additional wash and elution solvents were evaluated for the optimal methanol content. Additional washing was performed using 10-–40% methanol to remove more polar compounds than ginsenosides, and 60–100% methanol was found to elute the analytes efficiently (Fig. 4). The relatively polar ginsenosides, Rg1 and Re, tended to decrease the recovery in >30% methanol, and 100% methanol elution led to the highest recoveries of all compounds. Therefore, the selected optimal conditions for the additional wash and elution were 20% methanol and 100% methanol, respectively.

Extraction recovery of each ginsenoside spiked in the cell media under different SPE conditions: a the additional washing conditions and b the elution conditions. Extraction recoveries were calculated by comparing the peak area ratios of the extracted samples versus their non-extracted counterparts
Eight different solvent compositions were evaluated to select the appropriate reconstituted solvent; 100% mobile phase B showed the highest detectability in most ginsenosides and was selected for LC-MS analysis (data not shown).
Method validation
Method validation requires an evaluation of the linearity, LOD, LOQ, precision, and accuracy using the calibration samples prepared from the cell media and lysates. Calibration curves were generated for all analytes using the QC samples fortified with the reference standards at 10 different concentrations (0.1–200 ng/mL or ng/106 cells). Validation of the cell lysates was carried out using MCF-7 human breast cancer cells because the ginsenosides had different structure-related anticancer activities on those cells. The devised method was found to be linear (correlation coefficient, r 2 > 0.992) for all analytes (Table 1). Most compounds had an LOQ in the 0.5∼2.0 ng/mL range except for Rb1, PPT, and PPD, which has a LOQ of 5 ng/mL. The other relatively high LOQ of PPT and PPD might have originated from the low sensitivity due to the molecular properties not from cell matrix interference. The assay precision and accuracy were determined by analyzing three QC samples containing different concentrations of the individual ginsenosides (5, 20, and 100 ng/mL). The intra-day (n = 4) precision (expressed as %CV) ranged from 1.4% to 11.3%, while the accuracy (expressed as %bias) ranged from 95.9% to 106.2%. The inter-day (n = 4) precision (%CV) and accuracy (%bias) ranged from 3.1% to 11.6% and from 95.8% to 106.2%, respectively (Table 2).
For method validation in the cell culture media, the calibration curve was also linear (r 2 > 0.995) with similar LOD and LOQ levels to that of the cell lysates. However, no matrix effects were observed in the cell media. The intra- and inter-day variations ranged from 1.9% to 10.8% and 3.3% to 11.2%, respectively. The intra- and inter-day accuracies ranged from 94.9% to 105.4% and 96.3% to 106.4%, respectively (Table 2). The average extraction recovery ranged from 85% to 106% for all compounds analyzed at three different cellular concentrations in triplicate (see Electronic supplementary materials Fig. S2).
Quantitative determination for selective cellular uptake of ginsenosides
MCF-7 cells were incubated with either WG or RG for 6 or 12 h to determine the selective cellular uptake. The validated method was used to measure the ginsenoside concentrations from the cell culture media and cell lysates with each ginsenoside-enriched fraction from WG (10 and 50 μg/mL) and RG (10 and 50 μg/mL), which are composed of different ginsenosides. The treated concentrations were selected according to the concentration showing anticancer activities of RG (IC50 ∼ 37 μg/mL). However, WG did not exhibit anticancer activity at the same concentration. The content of ginsenosides from WG were as follows: 4.80% of Rg1, 6.04% of Re, 0.77% of Rf, 10.35% of Rb1, 4.02% of Rb2, 3.63% of Rc, 2.09% of Rd, and 0.89% of Rh1. RG contained 30.5% of Rg3, 7.63% of Rk1, 8.71% of Rg5, 1.27% of compound-K, 1.52% of Rh2, 0.17% of PPT, and 0.43% of PPD. Each sample (10 and 50 μg/mL of WG or RG) in the cell media was incubated without MCF-7 cells for 6 or 12 h to measure the stability of the ginsenosides (see Electronic supplementary materials Fig. S3). More than 92% of each ginsenoside remained in the cell media after 12 h, which showed that all ginsenosides were stable during the cellular uptake. According to the results, less polar and protopanaxadiol class ginsenosides, such as ginsenoside Rg3, Rk1, Rg5, Rh2, compound-K, and PPD, showed cellular uptake on the MCF-7 cells. However, relative polar and protopanaxatriol class ginsenosides did not accumulate in the cells (see Electronic supplementary materials Fig. S4).
The uptake ratio of each ginsenoside was determined by measuring the amounts of ginsenoside in the cell lysates from the treated amounts (Fig. 5). The PPD, which is an aglycone of protopanaxadiol type ginsenosides and has the most non-polar character, had the highest uptake ratio. This suggests that the preferential selective uptake of ginsenosides occurs according to their unique structure, which is in accordance with previous reports, which found that ginsenosides showed different anticancer activities as their structures [2, 5, 6, 9]. The uptake kinetics and subcellular distribution of ginsenosides would be addressed when they are accumulated in the cells using the present method.

Cellular uptake ratios of the ginsenosides by MCF-7 human breast cancer cells. The cancer cells were incubated with each ginsenoside mixture (from white and red ginsengs) at a concentration of 10 or 50 μg/mL and for 0, 6, and 12 h. The MCF-7 cells were washed, counted, and extracted. The ginsenosides accumulated in the cell were determined by a comparison to the standards, and then their uptake ratios were measured from the treated amounts
Conclusions
This study is a sensitive, selective, and validated LC-MS method that allows quantitative determination of the selective cellular uptake of ginsenosides in the cell lysates and culture media. Fifteen ginsenosides were separated using a reverse-phase 2-μm sub-particle size C18 column, and mass spectrometer detection was optimized for lower detection limits than reported previously. The sample preparation methods were also optimized to minimize the interference in the cell matrices and loss of ginsenosides during sample preparation. The devised method could be used to examine the behavior of ginsenosides on cancer cells and determine their associated structure-related anticancer activity.
References
Sengupta S, Toh SA, Sellers LA, Skepper JN, Koolwijk P, Leung HW, Yeung HW, Wong RN, Sasisekharan R, Fan TP (2004) Circulation 110:1219–1225
Yue PY, Mak NK, Cheng YK, Leung KW, Ng TB, Fan DT, Yeung HW, Wong RN (2007) Chin Med 2:6
Christensen LP (2009) Adv Food Nutr Res 55:1–99
Gao QG, Chen WF, Xie JX, Wong MS (2009) J Neurochem 109:1338–1347
Bae EA, Shin JE, Kim DH (2005) Biol Pharm Bull 28:1903–1908
Yim HW, Jong HS, Kim TY, Choi HH, Kim SG, Song SH, Kim J, Ko SG, Lee JW, Bang YJ (2005) Cancer Res 65:1952–1960
Wang CZ, Zhang B, Song WX, Wang A, Ni M, Luo X, Aung HH, Xie JT, Tong R, He TC, Yuan CS (2006) J Agric Food Chem 54:9936–9942
Yu Y, Zhou Q, Hang Y, Bu X, Jia W (2007) Cancer 109:2374–2382
Wang W, Zhao Y, Rayburn ER, Hill DL, Wang H, Zhang R (2007) Cancer Chemother Pharmacol 59:589–601
Serafim TL, Oliveira PJ, Sardao VA, Perkins E, Parke D, Holy J (2008) Cancer Chemother Pharmacol 61:1007–1018
Chen H, Ahn R, Van den Bossche J, Thompson DH, O'Halloran TV (2009) Mol Cancer Ther 8:1955–1963
Joo EJ, Ha YW, Shin H, Son SH, Kim YS (2009) Biol Pharm Bull 32:548–552
Li L, Sheng YX, Zhang JL, Wang SS, Guo DA (2006) Biomed Chromatogr 20:327–335
Li X, Sun J, Wang G, Hao H, Liang Y, Zheng Y, Yan B, Sheng L (2007) Biomed Chromatogr 21:735–746
Wang X, Zhao T, Gao X, Dan M, Zhou M, Jia W (2007) Anal Chim Acta 594:265–273
Xia C, Wang G, Sun J, Hao H, Xiong Y, Gu S, Shang L, Zheng C (2008) J Chromatogr B 862:72–78
Liu A, Pajkovic N, Pang Y, Zhu D, Calamini B, Mesecar AL, van Breemen RB (2006) Mol Cancer Ther 5:2879–2885
Tian Q, Failla ML, Bohn T, Schwartz SJ (2006) Rapid Commun Mass Spectrom 20:3056–3060
Gaspar JR, Qu J, Straubinger NL, Straubinger RM (2008) Analyst 133:1742–1748
Ha YW, Lim SS, Ha IJ, Na YC, Seo JJ, Shin H, Son SH, Kim YS (2007) J Chromatogr A 1151:37–44
Cho HJ, Kim JD, Lee WY, Chung BC, Choi MH (2009) Anal Chim Acta 632:101–108
Acknowledgments
This study was supported by an intramural grant from the Korean Institute of Science and Technology (KIST) and by grants from the National R&D Program of the Korean Ministry of Education, Science and Technology (MOEST) and the Korean Science and Engineering Foundation (KOSEF).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM 1
(PDF 989 kb)
Rights and permissions
About this article
Cite this article
Ha, Y.W., Ahn, K.S., Lee, JC. et al. Validated quantification for selective cellular uptake of ginsenosides on MCF-7 human breast cancer cells by liquid chromatography–mass spectrometry. Anal Bioanal Chem 396, 3017–3025 (2010). https://doi.org/10.1007/s00216-010-3515-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-3515-0