
Abstract
Current screening and event-specific polymerase chain reaction (PCR) assays for the detection and identification of genetically modified organisms (GMOs) in samples of unknown composition or for the detection of non-regulated GMOs have limitations, and alternative approaches are required. A transgenic DNA fingerprinting methodology using restriction enzyme digestion, adaptor ligation, and nested PCR was developed where individual GMOs are distinguished by the characteristic fingerprint pattern of the fragments generated. The inter-laboratory reproducibility of the amplified fragment sizes using different capillary electrophoresis platforms was compared, and reproducible patterns were obtained with an average difference in fragment size of 2.4 bp. DNA insert fingerprints for 12 different maize events, including two maize hybrids and one soy event, were generated that reflected the composition of the transgenic DNA constructs. Once produced, the fingerprint profiles were added to a database which can be readily exchanged and shared between laboratories. This approach should facilitate the process of GMO identification and characterization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic tests relying on screening polymerase chain reaction (PCR) assays followed by event-specific PCR assays are generally considered the most sensitive and specific methods for detection and identification of genetically modified organisms (GMOs) [1–3]. However, a major limitation of this approach is that a separate event-specific assay is required for each event. Event-specific methods for the detection of many known GMOs have been developed and extensively evaluated through inter-laboratory validations (http://gmo-crl.jrc.ec.europa.eu), but the unintentional release of non-regulated or unknown GMOs such as Bt10, LLRice601, and Bt 63 rice highlight the need for a capacity to rapidly identify unauthorized GMOs to limit their dissemination [4–6]. Taking into account the huge number of new genetically modified (GM) varieties in the pipeline, new generic identification approaches could also be an invaluable tool to both seed developers and regulators [7, 8].
PCR assays that target well-known sequences such as the inserted promoter or terminator elements in transgenic plants are the basis of GMO screening methods [9]. The advantage of screening assays is that a single screen can detect a number of GM events since any event containing the target element will generate a positive signal. However, such a GMO screen will produce an amplicon of the same size for all GM events containing the targeted DNA sequence. Therefore, additional testing by event-specific PCR assays is required for specific identification. Use of event-specific PCR is generally limited to GMO varieties that are approved or pending regulatory process because the sequences, constructs, and protocols are available, as they must be supplied to the regulatory agencies. Consequently, when a plant sample is positive in a screening PCR or in a GMO protein based assay, but negative to a panel of known event-specific PCRs, the presence of unapproved or unknown GMOs can only be indirectly suspected [7, 9, 10].
Different approaches such as identity tags, microarrays, and multiplexing have been promoted as alternative methods to identify GMOs [11–15]. Identity tags require international consensus, which would be difficult to apply without widespread adoption and might not be useful for products of unknown origin [16]. Some GMO microarrays such as CanoGMOChip® (Array-On GmbH, Gatersleben, Germany), NimbleExpress® arrays (Affymetrix, Santa Clara, CA, USA), and DualChip® GMO (Eppendorf, Namur, Belgium) are now commercially available [17]. However, for all of these methods, new or unknown genetic elements are particularly difficult to identify [17, 18]. In addition, since assay development for each new GM event requires additional primer and probe design and validation, the authentication of GMOs remains a major challenge [19, 20].
Several research teams have had success in characterizing the transgenic insertion site by fingerprinting the T-DNA junction or other transgenic sequence elements using genome-walking type PCR [21–26], providing information on the plant genome that flanks the transgenic sequence. Based on the work of Spertini et al. [22], we have developed and evaluated an application of this insert fingerprinting technique to detect and characterize the fingerprint pattern for individual GM events. By changing the orientation of the fingerprint toward the inserted gene instead of the plant genomic flanking region, the DNA insert fingerprint provides a characteristic pattern for the GMO construct that could be used to identify known and unknown inserted genes under the control of transgenic promoters and terminators. For the same primer set, the fingerprint pattern will vary due to differences in sequences, as restriction enzymes will cut at different positions in the gene insert. We have built a simple profile database targeting the 35S promoter (P35S) of the Cauliflower mosaic virus and the nopaline synthase terminator (TNOS) from Agrobacterium tumefaciens that could be used to identify at least 11 different GMOs. The reproducibility of the fingerprint profile was compared in a Canadian (CFIA) and an Australian (NMI) laboratory using two different electrophoresis platforms as well as different samples and reference materials. This methodology could represent a promising alternative for the detection and identification of GMOs, either approved or unapproved.
Materials and methods
Seeds and DNA extraction
Unless otherwise specified, all results were obtained using certified reference materials (ERM-BF410 to ERM-BF416d, ERM-BF424d) purchased from the Joint Research Centre—Institute for Reference Materials and Measurements (Geel, Belgium). MON88017, MON863xNK603, MON863xNK603xMON810, and T25 were purchased from the American Oil Chemists Society (Urbana, IL, USA). Genetically modified Roundup Ready GTS 40-30-2 soybean, Bt176 (N09K9, MAX40), and Bt11 maize (N44P4, 27M3) and their respective isolines (G4030, 09A4 and 26L6, N4424) were purchased from NK (St-Pie, Canada). Starlink seeds (CBH-351) were kindly provided by the Food and Drug Administration (USA). At CFIA, 10 g of seed was finely ground using a Foss Knifetec 1095 sample mill (Fisher). Genomic DNA was isolated from 1 g of seed powder using a DNeasy Plant extraction Kit (Qiagen Inc, Mississauga, Canada) according to manufacturer’s instructions, eluted, and quantified by UV spectrophotometry. At NMI, all reference materials were purchased pre-ground. Genomic DNA was isolated from 20 mg of powder using an adapted version of the Wizard Genomic DNA Purification Kit for isolation of Genomic DNA from Plant Tissue (Promega Corp, Madison, USA). Finely ground reference material (20 mg) was weighed into a microcentrifuge tube. Nuclei Lysis Solution (600 μL) was added, and the tube was vortexed for 30 s then incubated at 65 °C for 20 min. RNaseA (400 ng) was added, then incubated at 37 °C for 15 min before cooling the sample on ice for 5 min. Protein Precipitation Solution (250 μL) was added to the tube, vortexed vigorously for 20 s, then centrifuged at 14,000 rpm for 10 min. Supernatant (600 μL) was added to 600 μL of cold isopropanol in a clean microcentrifuge tube, inverted to mix, and centrifuged at 13,000 rpm for 5 min. The supernatant was decanted, and the pellet was washed with 600 μL of cold 70% ethanol and centrifuged at 13,000 rpm for 1 min, then the supernatant was decanted, and the pellet was air-dried for 20 min. The DNA was re-hydrated in 30 μL TE0.1 (10 mM Tris HCl pH 8.0, 0.1 M EDTA) overnight at 4 °C. Extracted DNA was evaluated for quality by gel electrophoresis and quantity by UV spectrophotometry and PicoGreen assay (Invitrogen).
DNA insert fingerprint
The DNA insert fingerprint method is schematically represented in Fig. 1. Briefly, 500 ng of genomic DNA was digested for 3 h with 40 U of TaqI (Invitrogen, Mississauga, Canada), HinP1I, or MspI (New England Biolabs, Mississauga, Canada) in a final volume of 50 μL using the temperature and the buffer recommended by the manufacturer. An adaptor was prepared by incubating 100 μM of both Upper and Lower adaptor strand oligonucleotides (see Table 1) at 80 °C for 2 min and decreasing this temperature 1 °C every 45 s down to 22 °C. The digested DNA (20 μL) was ligated for 1 h at 16 °C to 5 μL of 1.5 μM adaptor using 400 cohesive end ligation units of T4 DNA ligase (NEB) and 1× ligase buffer (NEB) in a final volume of 30 μL. The enzymes were inactivated by a 20-min incubation at 80 °C. A nested two-step PCR was used with an initial pre-selective PCR followed by a selective PCR. The pre-selective PCR was performed in 50 μL using 10 μL of the DNA-adaptor ligation reaction, 0.2 μM of adaptor primer 1, and 0.2 μM of P35S-specific primer 1. The cycling parameters for the pre-selective PCR were as follows: 2 min at 95 °C, then 34 cycles of 30 s at 95 °C and 3 min at 69 °C, followed by 4 min at 69 °C for the final extension. The selective PCR was performed in a 50 μL final volume with 1 μL of 100-fold dilution of the pre-selective PCR, 0.2 μM of adaptor primer 2, and 0.2 μM of P35S- or TNOS-labeled primer 2. WellRED dye-labeled primers (Sigma-Proligo) were used at CFIA, and 6-FAM primers (Operon) were used at NMI. The cycling parameters for the selective PCR were the same as for the pre-selective PCR except that only 29 cycles were used. In multiplex assays, 0.2 μM of both P35S-specific primer 1 and TNOS-specific primer 1 were used in the pre-selective PCR, 0.2 μM of both the P35S- and TNOS-labeled primer 2 were used in the selective PCR, and the annealing/elongation temperatures were 65 °C for both the pre-selective and selective PCR assays. All PCR assays were performed using 0.2 mM dNTPs, 1× Picomaxx Buffer, 2.5 U Picomaxx polymerase (Stratagene, La Jolla, CA, USA) on the GeneAmp 9700 (Applied Biosystems, Foster City, CA, USA) for CFIA, Canada, and on the Mastercycler ep Gradient S (Eppendorf, Germany) at NMI, Australia. Perfect Match PCR enhancer (1U) (Stratagene) was added in the selective PCR.

Overview of the technique used to obtain transgenic DNA insert fingerprint. Genomic DNA containing a transgenic DNA construct (a) is digested with restriction enzyme (b). Adapter units are ligated to cut ends of the DNA fragments (c), and this is followed by pre-selective amplification using the transgenic element-specific primer 1 and adapter-specific primer 1 (d). The first round of amplification can only be from the transgenic element-specific primer, as the adapter-specific primer binding site is complementary to the long single-strand of the adapter. Selective PCR using the transgenic element-specific labeled primer 2 and adapter-specific primer 2 (e) produces the fluorescently labeled amplicon, which is then analyzed by capillary electrophoresis (f). The theoretical fragment size is calculated by determining the length of the sequence from the 5′ end of the transgenic element-specific primer 2 binding site to the first base of the restriction enzyme binding site (inclusive) and then adding an additional 38 bp to account for the sequence through to the 3′ end of adapter-specific primer 2 binding site (g)
Capillary electrophoresis analysis
CEQ 8000 fragment analysis
At CFIA, the products of the selective PCR were purified using a QIAquick PCR purification kit (Qiagen), eluted using 30 μL 10 mM Tris–Cl pH 8.5, and diluted 1:20 in DNase/RNase-free distilled water (Gibco, Invitrogen). Of this diluted purified PCR, 5 μL was added to 35 μL of sample loading solution (SLS) (Beckman Coulter) together with the equivalent of 0.058 μL of 600 bp size standard (CEQ DNA Size Standard Kit 600, Beckman Coulter) and transferred into a 96-well plate. Each well was overlaid with a drop of mineral oil. Samples were analyzed by capillary electrophoresis in the CEQ 8000 system using a 33 cm × 75 μm capillary array following the manufacturer’s recommendations (Beckman Coulter, Ville Saint-Laurent, Canada). Sample migration took place under a capillary temperature of 35 °C. After DNA template denaturation at 90 °C for 120 s, samples were injected at 2 kV for 30 s, and fragment separation took place at 6 kV during a total of 60 min in two steps. The first step was performed at 6 kV with a ramp duration of 2 min followed by a second step at 6 kV with a departure time of 10 min and ramp duration of 5 min. The generated peaks were analyzed using Beckman fragment algorithm v2.2.1. The peak criteria parameter set included a slope threshold of 10, a relative peak height threshold of 5%, and a size estimation confidence of 95% using cubic model and PA v.1 dye mobility calibration. Electropherograms where size standards were missed more than three times or the size standard calibration deviation was greater than 1 bp were excluded from the analysis. Peaks auto-generated by electrical current variability or variability were also excluded. Samples giving signal above 130,000 relative fluorescence units (RFU) were diluted again 1:1 in SLS and reloaded. The negative control background threshold was defined using maize isoline maximum peak height plus three standard deviations (n = 3) between 100 and 600 bp. The detection limit (LOD) was set as the concentration where 50% of the tested samples gave a reproducible peak signal above the negative control background threshold. The peak height was calculated in RFU as provided by the fragment algorithm after the system dye spectra matrix correction.
ABI 3100 sequence analyzer
At NMI, the products of the selective PCR were diluted 1:30 in sterile, nuclease-free distilled water (Amresco). Of this diluted PCR product, 2 μL was added to 7 μL of Hi-Di™ Formamide (Applied Biosystems) plus 1 μL of 500 bp size standard (GeneScan™-500 ROX Size Standard, Applied Biosystems). The sample mix was denatured for 3 min at 95 °C on a GeneAmp9700 and kept on ice until further use. The sample mix was transferred to a 96-well plate, capped, and analyzed by capillary electrophoresis on the ABI 3100 Genetic Analyzer using a 36 cm × 75 μm, four-capillary array following the manufacturer’s recommendations (Applied Biosystems), using ABI Run 3100—Avant Data Collection v2.0 software. Sample migration took place with an oven temperature of 60 °C. After DNA template denaturation at 15 kV for 180 s, samples were injected at 3 kV for 5 s, and fragment separation took place at 15 kV in ten steps, during a total of 25 min. The generated peaks were analyzed using Gene Mapper v4.0 software, microsatellite analysis type, and the basic peak detection algorithm. The peak criteria parameter set included a homozygous minimum peak height signal level of 200, heterozygous minimum peak height level of 100, a minimum peak height ratio of 0.5, and a maximum peak width of 1.5 bp.
Electropherograms where the sizing quality was less than 0.75 were excluded from the analysis. Samples giving a peak height above 8,000 were re-diluted 1:50 from the products of the selective PCR as above and re-analyzed. The detection limit was set as the lowest transgene concentration where the tested samples gave a consistent and reproducible peak signal above the negative background threshold.
DNA sequencing
At CFIA, amplified DNA was analyzed by gel electrophoresis on a 1% agarose gel prepared with 0.89 M Tris, 0.89 M boric acid, and 20 mM EDTA (TBE) containing 0.5 μg/100 mL ethidium bromide for 1 h at a constant voltage of 100 V. DNA bands were visualized with a UV-transilluminator. Amplified DNA fragment bands obtained from maize events using the P35S-specific primers were excised and purified with a QIAquick PCR purification kit (Qiagen, Canada) and subsequently sent for sequencing (Plate-forme d'analyses biomoléculaires, Laval University, Canada) using P35S sequencing primer.
At NMI, samples identified by fingerprinting to have multiple amplified products were subjected to gel electrophoresis on a 2.5% agarose gel prepared with 1x TBE containing 0.5 μg/100 mL ethidium bromide for 1 h at a constant voltage of 100 V. DNA bands were visualized with a UV-transilluminator, excised, and purified with a QIAquick PCR purification kit (Qiagen, Australia). The purified product was diluted 1:10 and quantified by UV spectrophotometry. Purified product (5–50 ng), or the nested-PCR product in samples with only a single PCR product, was amplified with unlabeled P35S or TNOS sequencing primers (see Table 1) using the BDT Cycle Sequencing kit v3.1 as per manufacturer’s recommendations (Applied Biosystems) on the Mastercycler ep Gradient S. The extension products were purified with BigDye X Terminator Purification kit (Applied Biosystems) and analyzed on an ABI 3100 Genetic Analyzer using a 36 cm × 75 μm, four-capillary array following the manufacturer’s recommendations (Applied Biosystems), using ABI Run 3100—Avant Data Collection v2.0 software, and analyzed using Sequence Analysis Software v5.1.1.
The nucleic acid sequences obtained were compared with the National Center for Biotechnology Information GenBank sequence database and GMDD sequence database using the computer alignment software BLAST 2.2.19 [27, 29].
Results
Simplex and multiplex fingerprint analysis
In order to be characteristic of a GMO construct, a fingerprint profile has to be very specific and reproducible. Samples that do not contain GMO material should not generate a profile, while events that contain the same construct should generate the same peak patterns. Thirteen positive and five negative reference materials were tested on different instrument platforms in two different countries. In addition, two GMOs (Bt11 and Bt176) were tested using both reference materials and high purity commercial samples in addition to their respective negative isolines. Only transgenic events containing the targeted promoter P35S or terminator TNOS sequences were amplified to produce a fingerprint profile after the nested PCR (Fig. 2). Non-transgenic isolines (e.g., N4424, 09A4, N26L6, and G4030), which differ only from their GMO counterpart by the absence of the transgene, did not produce any significant fragment signal above 1,000 RFU for P35S fragments and 500 RFU for TNOS fragments using the CEQ 8000 fragment analysis conditions. Analysis of genomic DNA from GM events, including Bt11, Bt176, and MON810 maize and GTS 40-3-2 soybean resulted in reproducible electropherogram patterns between 80 and 640 bp. The profiles produced using a P35S or TNOS simplex approach were compared to the profiles obtained using a P35S and TNOS multiplex amplification. The same characteristic fragment patterns were detected although the maximum peak height was decreased in multiplex (Fig. 3). Bt11 reference materials, as well as the Bt11 varieties N44P4 and 27M3, shared the same peak pattern (Electronic supplementary material). Between assays, the intra-laboratory variation in position of the primary peak of Bt11 was less than 0.5 bp. Similar observations were noted with Bt176.

Typical DNA insert fingerprint electropherogram results. Multiplex assay profile obtained by skipping the pre-selective PCR amplification and using only the selective PCR run on 1% Bt11 (A). Multiplex assay profile obtained following nested multiplex PCR on non-GM maize (B) and 1% Bt11 (C). These profiles were obtained using HinP1I digestion and the CEQ 8000. The P35S- and TNOS-labeled fragments signal are represented in blue and green, respectively, while the size marker signal is red

Serial dilution curves analyzed by DNA insert fingerprinting. P35S and TNOS maximum peak height were obtained using MspI with GM maize events Bt11 (N44P4) DNA diluted with its isoline DNA (N4424) and Bt176 (MAX40) DNA diluted with its isoline DNA (G4030) either in simplex (S) or multiplex (M) (n = 3). The fingerprint and the data analysis were performed using the CEQ 8000
For instance, Bt11 tested with P35S primers generated two detectable fragments with both TaqI and MspI and a single fragment with HinP1I. Bt11 contains one copy each of the cry1Ab and the pat genes both regulated by their own 35S promoter [28] (Table 2), which would account for the two fragments observed when using both the TaqI and MspI restriction enzymes. Since only a single fragment was observed using the HinP1I restriction enzyme, it is likely that the second HinP1I-generated fragment did not fall within the 80 to 640 bp capillary electrophoresis analysis window and was thus not detected using this fingerprinting approach. Indeed, according to the Bt11 sequence available through the GMDD database [29], HinP1I cuts at position 2525 inside cry1Ab, generating a fragment of 1,176 bp using the P35S-specific primers.
On some occasions, small secondary peaks less than 10% the height of the major peaks were apparent at higher GM concentrations. For instance, a small peak was detected using TNOS primers at 314 bp in maize samples with 1% Bt11 and above (Fig. 2 C). These secondary peaks had no impact on the identification of GM biomarker fragments since their signal was only a fraction of the size of the major peak present in the fingerprint. Consequently, only the reproducible major peaks were compared and added to the database. Small variations in the target sequence resulting in lower affinity for the primers or incomplete digestion of the genomic DNA could account for the occasional presence of minor fragments. Although some fragments were weakly amplified with the use of the selective PCR run only (Fig. 2 A), no reproducible patterns were obtained without the use of a full nested PCR.
Sensitivity
The detection limit was below 1% GMO content (w/w) using the current protocol. However, the maximum peak height did vary substantially depending on the event being analyzed and the primer set used (Fig. 3). For instance, using P35S primers the maximum peak height following analysis of 1% (w/w) Bt11 was almost double the peak height obtained following analysis of 1% (w/w) Bt176. In some samples, such as Bt11 when tested with TNOS primers and HinP1I, the detection level was as low as 0.1% (w/w) (data not shown). Multiple inserts of the same construct into a GMO will result in multiple copies of the same P35S and TNOS target sequences for fingerprint generation if these sequences are present in the construct. In addition, primer affinity may vary between events if there is any sequence variation within the critical primer binding regions for the pre-selective and selective PCR assays. Either of these factors could affect the observed peak height in the fingerprint data. Above a 1–5% (w/w) GMO concentration, the fragment amplification appears to plateau, and no correlation between GM concentration and peak height was observed. This could be expected from a nested PCR given that this technique is only qualitative. With 100% GM seeds, the dye signal was well above the instrumentation limits, and the nested PCR product had to be diluted and reloaded. This dilution was necessary to avoid capillary overload and removed small non-specific fragment signals from the main fragment profile. The position of the primary peak did not vary with the GMO concentration.
GMO fingerprint database
The fragment patterns were entered into a DNA insert fingerprint database to identify GMOs (Table 2). DNA insert fingerprints for 12 different maize events, including two maize hybrids and one soy event, were generated that reflected the composition of the transgenic DNA constructs. Based on the specific sequence recognized by the restriction enzymes, generated fragments differed between most transgenic events. A notable exception was MON810 and NK603, which could not be differentiated by P35S generated fragments. In both events, the P35S regulating sequence is flanked by a maize heat-shock protein 70 sequence element of ∼800 bp [31]. On the other hand, during integration of the MON810 event, the NOS terminator was lost due to 3′ truncation [28]. Since NK603 but not MON810 contains NOS sequences, these GMO were differentiated by the presence of detectable amplicons in NK603 when restriction enzyme fragments from TaqI and MspI were amplified by the TNOS primer set. When neither the P35S promoter nor the TNOS terminator was present in a GM construct, a fingerprint pattern was not generated using these specific target primers. Nonetheless, in some cases, such GM constructs were detected using a primer set specific for another target sequence. For instance, GM canola event GT73 was detected using primers specific for the figwort mosaic virus promoter (data not shown). Each GM event tested did have a unique fingerprint pattern with the exception of GMOs with stacked genes. The fragment pattern generated by the stacked event MON863xNK603xMON810 was the same as the combination of the results for the non-stacked events MON810, NK603, and MON863.
Where possible, amplified fragments were sequenced to confirm their identity (Electronic supplementary material). In addition, their sequence was compared with those for the expected sequence theoretical fragment size. The expected sequence was assembled by joining the relevant selective primer sequence with the relevant sequences from the GenBank database, if available, and the adaptor selective primer sequences (Fig. 1g).
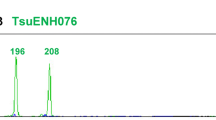
Sequencing data indicated a close match between the reported GenBank sequence and the sequences from the fingerprint of the T25 maize event (Fig. 4). However, both the sequences and the amplicons are four and six bases, respectively, smaller than the theoretical fragment size. The observed P35S fragment sizes for the T25 maize event HinP1I and MspI fingerprints using the ABI 3100 instrument were 489 and 120 bp, respectively. The observed values for the TaqI fingerprint using the CEQ 8000 and ABI 3100 instruments were 137 and 138 bp. We have not found an explanation for the slightly larger variation observed with T25 fragments, which was observed with few events (Table 2). Although the variation could be the result of a difference between the reported sequence and the event or the variety tested, it could be also the results of a “DNA polymerase slip” due to the nested PCR.

Sequence alignment of T25 maize P35S fingerprints generated using HinP1I, TaqI, and MspI. Fingerprint fragments produced using the P35S primers set are aligned to AY629235.1 and DQ156557.1 both from Zea mays transgenic phosphinothricin acetyltransferase genes. The red underline is the P35S promoter-specific primer 2 binding site. The green dotted underlines indicate the restriction enzyme binding sites. The blue underline is the 5′ end of the upper adapter strand sequence to the 3′ end of the adapter primer 2 binding site
In the absence of relevant DNA sequences attributed to a GM event in the GenBank database, DNA sequence analysis still provided information on the junction between either the P35S or TNOS target sequence and the adjacent transgenic DNA sequences. For example, the P35S-amplified HinP1I-based fingerprint from MON88017 produced a fragment of 341 bp (Table 2). While the relevant MON88017 event sequences are not available in the GenBank database, a BLAST search revealed that the 341 bp fragment comprised a portion of the Cauliflower mosaic virus 35S promoter (96% identity with 66 bp from GenBank Accession AY592594) adjacent to sequences matching the 5′ untranslated leader from the wheat major chlorophyll a/b-binding protein (80% identity with 39 bp from GenBank Accession M10144) with further downstream sequences matching the rice actin gene (95% identity with 119 bp from GenBank Accession EU155408). This analysis is consistent with the MON88017 expression cassette for the cry3Bb1 gene according to Agbios [28]. Interestingly a 237 bp MspI fragment initially not detected was theoretically predicted based on additional MON88017 sequence data in the GMDD database [29]. MON88017 was retested, and a fragment was detected at 236 bp, suggesting that an inefficient digestion during the initial testing might have prevented its detection. Routine application of the DNA insert fingerprinting technique may require the development of positive external or internal controls to be included in each run since false-negative results could be difficult to identify.
Reproducibility and robustness
The reproducibility and robustness of the method was studied using two different capillary electrophoresis platforms in separate laboratories and different reference material lots in a collaborative study between the CFIA, Canada and the NMI, Australia (Table 2 and Fig. 5). For most fragments where theoretical data were available to enable a comparison, the fragment length tended to be slightly overestimated by the CEQ 8000 system (1.3 ± 4.2 bp) and was slightly underestimated by the ABI 3100 system (−2.1 ± 3.5 bp). The fingerprint profiles differed by an average of 2.4 bp between the CEQ 8000 and ABI 3100 systems. TNOS fragment variations (0.5 ± 1.0 bp) were smaller than the P35S fragment variations (3.1 ± 2.8 bp), suggesting some sequence or dye dependant factor. This later variation between CEQ and ABI system is similar (5.8 ± 4.6 bp) to that obtained using the ABI 3100 in another CFIA laboratory, testing the events Bt11, Bt176, CHB-351, T25/T14, and MON863 (Electronic supplementary material). While additional terminal A residues, which can be added by Taq polymerase, were observed during DNA sequence analysis and would result in a fragment up to two bases longer than the theoretical length, this does not completely account for the difference in theoretical and observed fragment lengths. A similar underestimation in fragment size has been reported by NMI previously using the ABI 3100 system [30] and is possibly due to differences in migration rate of the standard and the sample through the capillary electrophoresis system due to the different fluorescently labeled dyes attached to the standard and the sample. For instance, the fluorescent standards for the ABI 3100 systems utilized a ROX dye, while the DNA samples were labeled with a FAM dye, which could thus account for this discrepancy.

Inter-laboratory variation in fingerprint size. Three GMOs, Bt11, MON863xNK603, and T25, were tested in both laboratories using TaqI, MspI, and HinP1I, and the difference in size for each fragment between the CEQ 8000 and the ABI 3100 is reported relative to their size. Size variations are represented by closed circles for P35S and open circles for TNOS
Discussion
Event-specific PCR is a more rapid and economical methodology than the proposed DNA insert fingerprint to identify a GMO present in a sample when the sample composition is known or suspected. However, when the sample composition is unknown, it is neither practical nor feasible to analyze the sample with multiple event-specific assays covering a range of GM events. Under these circumstances, untargeted or multiple screening methods are more advantageous. Genome walking or anchored PCR T-DNA fingerprint analysis has been successfully used in the past to discriminate different T-DNA inserts in stably transformed plants [22, 23, 25], and these methods require less DNA, time, and work input than Southern blotting. The use of capillary electrophoresis increases the size determination accuracy of these approaches and allows effective use of the fingerprinting method for characterizing GM events. However, in order to rapidly identify multiple GM events, the fingerprints need to be oriented toward the inserted gene instead of the plant genomic flanking region (Fig. 1). Three reasons explain this choice of pattern orientation. Firstly, while GM seed developers may modify regulatory sequences such as the P35S and NOS sequences before introducing them into GM constructs, they tend to maintain the proximal regions of the regulatory sequences intact since the secondary structure of these sequences has most impact on regulation of gene expression. Hence, P35S or NOS-specific primers designed against the proximal region of these regulatory sequences will have the widest coverage in terms of detecting GMOs using the DNA fingerprint technique. Secondly, as the fragment size that can be analyzed using capillary electrophoresis is limited to a maximum of approximately 600 bp, having the region that is targeted by the assay close to the primer target region is important. Finally, while this orientation does not allow characterization of the GM event through profiling the junction between the transgenic insert and the plant genomic sequences, it provides information on the inserted gene or adjacent transgenic sequences. This information comes from the fingerprint pattern, which is characteristic of the insert construct and also from the insert DNA sequence. These transgenic DNA sequences can also be amplified and sequenced, which is a major advantage of the genome-walking-based methods [22].
The primer and restriction enzymes used are key elements in a DNA insert fingerprint. The selection of the promoter and terminator targets is important since only the GM events with those target sequences will be amplified and detected. Although P35S is the most frequently used promoter in approved GM maize according to the biotech crop databases Agbios and BATS, this promoter is not present in every transgenic crop [28, 31]. The use of a multiplex assay enables targeting of multiple regulatory elements and increases the probability of detecting different GMOs. The second target of the multiplex assay in this study is the TNOS terminator. TNOS is one of the most frequently used transgenic terminators. In Canada, 21 of the 23 approved GM maize events contain at least one of these genetic elements [28], while 12 of the 13 GM maize events approved by Food Standards Australia New Zealand for use in food in Australia contain at least one of these genetic elements. The choice of restriction enzymes is another component of the fingerprinting method that increases the probability of differentiating between GM constructs. The 4 bp recognition motif restriction enzymes MspI, HinP1I, and TaqI were used for the digestion of genomic DNA in order to increase the cleaving frequency and to produce fragment sizes in the range of the capillary electrophoresis analysis. The use of two or three enzymes increases the discrimination efficiency of a procedure that, otherwise, presents limited polymorphisms (the number of possible patterns). For instance, a single enzyme producing a single fragment with a capillary electrophoresis resolution of ±1 bp could generate only 150 unique profiles taking into account the non-template terminal A (600 bp capillary electrophoresis range/4 bp = 150 profiles). On the other hand, the use of three restriction enzymes increases the number of theoretical profiles to approximately 1503 or 3.3 × 106 combinations. The restriction endonucleases selected are isochizomers that generate the same overhang end and so are all complementary to the same adaptor sequence. However, other restriction enzymes with appropriately designed adaptor sequences could be used, with limited requirement for extra resources, since all reactions are performed in parallel. In comparison, multiplex assays for single nucleotide polymorphisms can simultaneously detect up to 48 short DNA sequences in GMOs but this approach does not allow direct sequence confirmation, which is an advantage of the DNA insert fingerprint and other genome-walking-type approaches [32].
Another element that was integrated into this methodology was the nested PCR. The use of a nested PCR increases the analytical specificity and sensitivity, decreasing the sample processing time and providing a much faster and direct characterization of the GMO than a procedure such as genome walking, which can require cloning of the fragment [21, 25, 26]. This insert fingerprint approach allows automation, and multiple samples can be tested in parallel. Another group has reported detection of a single integration site in a single cell in a background of 25 ng of salmon and soy DNA by anchored nested PCR following biotin–streptavidin isolation [21]. In this regard, optimization of the multiplex PCR primer set and the conditions of the DNA insert fingerprint method could decrease the detection limit to close to 0.1% (w/w). While a nested PCR can be more prone to cross-contamination and false-positive amplification, the fingerprint patterns are based on multiplex assays and are the result of different enzyme profiles tested in parallel, so cross-contamination after digestion is unlikely to reproduce a full fingerprint pattern. Nevertheless, replicate analysis and confirmatory methods such as DNA sequencing are required. When the adaptor or part of the targeted genetic element is identified by DNA sequencing, adjacent DNA sequences can then be used to confirm the identity of the amplified product. At least 24 fragments from the nested PCR were sequenced successfully (Electronic supplementary material). Theoretically, while the fingerprint pattern could not differentiate a stacked event from a seed mixture of non-stacked events, differentiation should be possible if the analysis is based on a single seed DNA extract. In a number of cases, the relevant GM event DNA sequence is not reported in the GenBank database. Under these circumstances, it is still often possible to characterize the transgene adjacent to the regulatory target sequence through GenBank searches. One promising application of this new approach is that fragment size information could be shared between laboratories. We have shown the reproducibility of the pattern using two capillary electrophoresis platforms. In fact, the database information is compatible with any high-resolution gel analysis and could be useful even using different anchored PCR approaches that target the same regulating sequence and use the same restriction enzyme. In that case, the only information that is needed to transpose the fragment size patterns to another method is simply the relative position of the target primer site. Sharing the GM DNA insert fingerprint size information could become a very effective way to rapidly identify GM constructs without disclosing any proprietary information. Once standardized for the user’s in-house laboratory settings, it could reduce the time dedicated to event-specific method acquisition or adaptation since the same methodology is applicable to multiple events. The comparison between experimental peak patterns and the GM database would allow a laboratory to make a preliminary identification of a GM event even without the reference material for the identified specific event. Peak patterns not found in the fingerprint database could be the result of newly approved GM events, point mutations or other variations of approved GM events, or the result of an unknown GMO. As described above, peaks corresponding to unknown fragments could be sequenced to further assess their origin or to alleviate concerns that they might be associated with an unknown product. Even though two transgenic constructs that share the same inserted gene and regulating elements may not be differentiated, the risk assessment on those genes might be available and provide valuable information. It would then be possible to make a risk-based analysis on those products.
Conclusion
DNA insert fingerprints were obtained for all the 12 GM maize events tested that contained either the P35S or TNOS sequence elements. A unique fragment profile was generated for the majority of the events. However, the identification of stacked events still remains a challenge. The extension of the application of the method to additional GM events would always be limited by the target sequences used in the multiplex nested PCR. Since about 90% of currently approved GMOs contain either the P35S or TNOS target sequence, there is still a long list of other GM events that could be detected using this approach and new DNA insert fingerprint pattern to be generated. Most importantly, the database of DNA insert fingerprint patterns is compatible with different detection platforms. Therefore, the DNA insert fingerprint provides an additional tool to detect GMOs and enables additional characterization, which is not possible using the current screening approach. It will assist in the rapid identification of GMOs in samples of unknown composition and could be used to detect unapproved GM events.
References
Garcia-Canas V, Cifuentes A, Gonzalez R (2004) Crit Rev Food Sci 44:425–436
Ahmed FE (2002) Trends Biotechnol 20:215–223
Holst-Jensen A, Rønning SB, Løvseth A, Berdal KG (2003) Anal Bioanal Chem 375:985–993
Akiyama H, Sasaki N, Sakata K, Ohmori K, Toyota A, Kikuchi Y, Watanabe T, Furui S, Kitta K, Maitani T (2007) J Agric Food Chem 55:5942–5947
Watanabe T, Shiramasa Y, Furui S, Kitta K, Minegishi Y, Akiyama H, Maitani T (2007) J Food Hyg Soc Jpn 48:170–178
Macilwain C (2005) Nature 434:423
Aarts HJ, van Rie JP, Kok EJ (2002) Expert Rev Mol Diagn 2:69–76
Anklam E, Neumann DA (2002) J AOAC Int 85:754–756
Hubner P, Studer E, Luthy J (1999) Nat Biotechnol 17:1137–1138
Lipp M, Brodmann P, Pietsch K, Pauwels J, Anklam E (1999) J AOAC Int 82:923–928
James D, Schmidt AM, Wall E, Green M, Masri S (2003) J Agric Food Chem 51:5829–5834
Kok EJ, Aarts HJ, Van Hoef AM, Kuiper HA (2002) J AOAC Int 85:797–800
Marillonnet S, Klimyuk V, Gleba Y (2003) Nat Biotechnol 21:224–226
Marillonnet S, Klimyuk V, Gleba Y (2003) Nat Biotechnol 21:974–975
Schmidt AM, Sahota R, Pope DS, Lawrence TS, Belton MP, Rott ME (2008) J Agric Food Chem 56:6791–6800
Pauli U (2003) Nat Biotechnol 21:598
Hamels S, Glouden T, Gillard K, Mazzara M, Debode F, Foti N, Sneyers M, Nuez TE, Pla M, Berben G, Moens W, Bertheau Y, Audqon C, Van Den Eede G, Remacle J (2009) Eur Food Res Technol 228:531–541
Leimanis S, Hamels S, Nazé F, Mbella GM, Sneyers M, Hochegger R, Broll H, Roth L, Dallmann K, Micsinai A, La Paz JL, Pla M, nen-Nieweler C, Papazova N, Taverniers I, Hess N, Kirschneit B, Bertheau Y, Audeon C, Laval V, Busch U, Pecoraro S, Neumann K, Sel S, Van Dijk J, Kok E, Bellocchi G, Foti N, Mazzara M, Moens W, Remacle J, Van Den Eede G (2008) Eur Food Res Technol 227:1621–1632
Xu J, Zhu S, Miao H, Huang W, Qiu M, Huang Y, Fu X, Li Y (2007) J Agric Food Chem 55:5575–5579
Onishi M, Matsuoka T, Kodama T, Kashiwaba K, Futo S, Akiyama H, Maitani T, Furui S, Oguchi T, Hino A (2005) J Agric Food Chem 53:9713–9721
Nielsen CR, Berdal KG, Holst-Jensen A (2008) Eur Food Res Technol 226:949–956
Spertini D, Beliveau C, Bellemare G (1999) Biotechniques 27:308–314
Pérez-Hernández JB, Swennen R, Sági L (2006) Transgenic Res 15:139–150
Côté MJ, Meldrum AJ, Raymond P, Dollard C (2005) J Agric Food Chem 53:6691–6696
Theuns I, Windels P, De Buck S, Depicker A, Van Bockstaele EV, De Loose M (2002) Euphytica 123:75–84
Akritidis P, Pasentsis K, Tsftaris AS, Mylona PV, Polidoros AN (2008) Electronic J Biotechnol 11:1–199
Zhang Z, Schwartz S, Wagner L, Miller W (2000) J Comput Biol 7:203–214
GM Crop Database, Agbios. Merrickville, Canada. http://www.agbios.com. Accessed 1 June 2009
Dong W, Yang L, Shen K, Kim B, Kleter GA, Marvin HJ, Guo R, Liang W, Zhang D (2008) BMC Bioinformatics 9:260
Partis L, Burns M, Chiba K, Corbisier P, Gancberg D, Holden MJ, Wang J, Liu QY, Okunishi T, Yang I, Vonsky M, Emslie KR (2007) Electrophoresis 28:3193–3200
GMO-Watch Report: Genetically Modified Crops: molecular and regulatory facts. BATS. Centre for Biosafety and Sustainability, Zurich, Switzerland. http://www.bats.ch/gmo-watch/. Accessed 1 June 2009
Chaouachi M, Chupeau G, Berard A, Mckhann H, Romaniuk M, Giancola S, Laval V, Bertheau Y, Brunel D (2008) J Agric Food Chem 56:11596–11606
Acknowledgments
Work conducted at the National Measurement Institute, Australia was funded by the Office of the Gene Technology Regulator, Australia. CFIA research was funded by the Canadian Regulatory System for Biotechnology (CRSB). We thank Kate Griffiths, Carole Simard, and André Perron for their constructive review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Kim L. Dibley and Somanath Bhat contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 408 kb)
Rights and permissions
About this article
Cite this article
Raymond, P., Gendron, L., Khalf, M. et al. Detection and identification of multiple genetically modified events using DNA insert fingerprinting. Anal Bioanal Chem 396, 2091–2102 (2010). https://doi.org/10.1007/s00216-009-3295-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-3295-6