
Abstract
We have developed and validated a quantitative liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI MS/MS) procedure for the simultaneous determination of seven natural and semisynthetic tropane alkaloids in plasma: atropine (d-hyoscyamine/l-hyoscyamine), cocaine, homatropine, ipratropium, littorine, N-butylscopolamine, and scopolamine. Plasma and serum samples were precipitated for deproteinization (recovery 88–94%), followed by reversed-phase-based liquid chromatography prior to positive electrospray ionization for detection by multiple reaction monitoring using a linear ion trap quadrupole mass spectrometer. All analytes were quantified using cocaine-d3 as an internal standard suitable and reliable for robust, precise (coefficient of variation 2–13%), and accurate (87–122%) measurement within a linear range of 3 orders of magnitude (0.05–50 ng/ml plasma). The method was exemplarily applied to stability studies in phosphate-buffered saline, human serum, and rabbit serum. Each alkaloid was incubated separately and samples were taken at distinct incubation time points. Supernatants of diverse alkaloids at corresponding time points were pooled and subjected to simultaneous LC-ESI MS/MS quantification. This combinatorial analysis design allowed us to analyze the stability of samples with a drastically reduced number of chromatographic runs. In the presence of rabbit serum, all tropane alkaloids tested were degraded significantly within minutes to hours, with the exception of the stable semisynthetic compounds ipratropium and N-butylscopolamine. In contrast, in the presence of equal concentrations of human serum, no degradation was observed for any of the compounds, with the exception of cocaine. Relevant enzymes involved in enzymatic degradation are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tropane alkaloids represent a class of bioactive small molecules including, e.g., littorine (Fig. 1f), its positional isomer l-hyoscyamine (Fig. 1a), atropine (racemic d-hyoscyamine/l-hyoscyamine, Fig. 1a), and l-scopolamine (hyoscine, Fig. 1h), which are biosynthesized in flowering plants of the Solanaceae family, e.g., Atropa belladonna (deadly nightshade), Datura stramonium (thorn apple), and Hyoscyamus niger (henbane) [1]. Most common natural tropane alkaloids as well as their semisynthetic pharmaceutical derivatives, e.g., homatropine (Fig. 1b), ipratropium (Fig. 1e), and N-butylscopolamine (Fig. 1g), act as antagonists of muscarinic receptors (antimuscarinic agents), thus affecting acetylcholine-mediated signal transduction in the organism [2, 3]. Owing to this anticholinergic activity in the central and peripheral system, antimuscarinic agents are used as therapeutics in, e.g., anesthesia (atropine), nausea (scopolamine), bronchitis and asthma (ipratropium), gastrospasm (N-butylscopolamine), and heart and eye disorders (homatropine) and as an essential antidote (atropine) for the treatment of poisoning by organophosphorus compounds (e.g., numerous pesticides and nerve agents) [3, 4]. Despite these beneficial effects, tropane alkaloids themselves are toxic in a dose-dependent manner, ultimately leading to death by respiratory paralysis [3]. Optimized therapeutic dosing is needed when administering these drugs to prevent the patient from suffering serious adverse effects. Therefore, a quantitative bioanalytical tool is mandatory for, e.g., drug monitoring, pharmacokinetics, stability studies, and forensic samples. Diverse techniques especially for atropine were introduced, including radioimmunoassays [5, 6], radioreceptor assays [7, 8], as well as separations by capillary electrophoresis [9, 10], thin-layer chromatography [11, 12], gas chromatography-mass spectrometry (MS) [13, 14], and liquid chromatography (LC) coupled with UV [15–17], fluorescence [18], conductometric [19], or MS [20–25] detection. LC-MS and LC-MS/MS techniques represent the most promising techniques as they overcome the drawbacks of the more traditional methods, e.g., (1) radioimmunoassays are of insufficient assay selectivity owing to cross-reactivity to structural analogs, (2) functional radioreceptor assays are time-consuming in terms of preparation and measurement and analyze the total competitive binding to muscarinic receptors (atropine equivalents) and thus do not allow one to discriminate single alkaloids, and (3) LC-UV detection procedures are of very limited sensitivity especially when complex biological matrices are analyzed. In addition, the benefits of combining high chromatographic resolution with highly selective mass detection make LC-MS/MS predestinated for multicomponent tropane alkaloid analysis. However, only a limited number of procedures for the detection of, e.g., atropine in the presence of scopolamine in plant extracts [22, 24–26] or its metabolites in rat plasma [20] have been reported in the literature so far. Therefore, we developed and validated a multicomponent method that enables measurement of atropine, cocaine, homatropine, ipratropium, littorine, N-butylscopolamine, and scopolamine. This procedure was applied to stability studies. To the best of our knowledge, no method has been described before allowing simultaneous quantification in plasma of the selected alkaloids with pharmaceutical, toxicological, and forensic relevance.

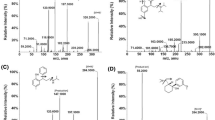
Tandem mass spectrometry (MS/MS) spectra of tropane alkaloids generated by electrospray ionization (ESI) and collision-induced dissociation: a atropine (d-hyoscyamine/l-hyoscyamine); b homatropine; c cocaine; d cocaine-d 3; e ipratropium; f littorine; g N-butylscopolamine; h scopolamine. Fragment spectra were obtained from tropane alkaloids dissolved in 0.1% (v/v) 70:30 (v/v) formic acid (FA)/acetonitrile (ACN) infused with positive ESI and dissociated by collision with nitrogen in a triple-quadrupole machine (API 4000 QTrap). Cleavage sites (dotted lines) were deduced and assigned in analogy to the fragments described before for atropine [20, 21, 24] and scopolamine [29]
Materials and methods
Chemicals
Acetonitrile (ACN; gradient grade), formic acid (FA; Uvasol), and all salts (guaranteed reagent) used for phosphate-buffered saline (PBS; 46 mM NaCl, 0.89 mM KCl, 4 mM KH2PO4, 4 mM Na2HPO4, pH 7.6) were purchased from Merck (Darmstadt, Germany). Atropine (CAS no. 51-55-8), l-hyoscyamine (CAS no. 101-31-5), l-scopolamine (CAS no. 51-34-3), N-butylscopolamine bromide (CAS no. 149-64-4), ipratropium bromide (CAS no. 60205-81-4), cocaine (CAS no. 50-36-2), and cocaine-d 3 (CAS no. 65266-73-1) were purchased from Sigma (St. Louis, MI, USA). Homatropine (CAS no. 78-00-3) was supplied by ABCR (Karlsruhe, Germany). Synthetic littorine (CAS no. 23018-08-8) was a friendly gift from Koichiro Shimomura (School of Life Sciences, Toyo University, Gunma, Japan). All alkaloids were supplied in a purity of 98% or better (thin-layer chromatography). Serum from individual White New Zealand rabbits was supplied by Harlan France (Gannat, France). Human EDTA plasma and serum were generated from blood of healthy donors using the corresponding types of Monovette from Sarstedt (Nürnbrecht, Germany).
For sample dilution purposes, a mixture of high performance LC (HPLC) eluent A and eluent B (80:20 v/v, for the composition, see leter) was prepared (80:20 mix).
LC–electrospray ionization MS/MS analysis
The binary LC system (two pumps, autosampler, column oven, and controller, all PE 200 series, PerkinElmer, Rodgau-Jügesheim, Germany) was coupled to an electrospray ionization (ESI) mass spectrometer controlled by the accompanying software: Analyst 1.4.2 (API 4000 QTrap, Applied Biosystems, Darmstadt, Germany). Chromatography was performed on an Atlantis T3 C18 column, 5 µm, 150 mm × 4.6-mm inner diameter (Waters, Eschborn, Germany), protected by a 5-µm poly(ether ether ketone)/polytetrafluoroethylene filter (Chromatographie-Handel Müller, Fridolfing, Germany). Eluent A was 0.1% (v/v) FA in water and eluent B was a mixture of 80:20 ACN/water and 0.1% (v/v) FA. Separations of 100-µl injection volume were carried out at 30 °C and 1 ml/min in gradient mode—time (min)/eluent B (%): 0/23; 5/38; 6/80; 7/80; 7.5/23; 8.5/23. MS detection was performed in the positive multiple reaction monitoring (MRM) mode using the following settings: ionization spray voltage 4,800 V, curtain gas 25 psi, heater gas 70 psi, turbo ion spray gas 60 psi, gas temperature 700 °C, entrance potential 10 V, and dwell time 50 ms. The gas pressure (nitrogen) for collision-activated dissociation was adjusted to a medium setting. Cocaine-d 3 was used as an internal standard for all analytes. The following individual settings for the transition, collision energy (CE), declustering potential (DP), and collision cell exit potential (CXP) were used for selective detection: atropine and l-hyoscyamine 290.3→124.3 (CE 35 V, DP 76 V, CXP 6 V), cocaine 304.2→182.2 (CE 29 V, DP 61 V, CXP 10 V), cocaine-d 3 307.2→185.2 (CE 29 V, DP 61 V, CXP 12 V), homatropine 276.3→124.1 (CE 33 V, DP 41 V, CXP 6 V), ipratropium 332.2→124.2 (CE 45 V, DP 111 V, CXP 6 V), littorine 290.3→124.3 (CE 35 V, DP 81 V, CXP 6 V), N-butylscopolamine 360.2→121.1 (CE 39 V, DP 71 V, CXP 8 V), and scopolamine 304.3→138.2 (CE 29 V, DP 61 V, CXP 8 V).
Preparation of alkaloid stock solutions
Tropane alkaloids were dissolved in methanol, resulting in concentrations of either 2 or 5 mg/ml. Further dilutions used for calibration standards in plasma (1 µg/ml) or spiking incubation mixtures for stability studies (10 µg/ml) were performed in the 80:20 mix.
Cocaine-d 3 (internal standard) was purchased as a methanolic solution (100 µg/ml), which was first diluted with the 80:20 mix (100 ng/ml) and finally mixed with eluent A to result in a concentration of 0.15 ng/ml (eluent A–internal standard mix).
Preparation of plasma samples and standards
Samples and blank
Plasma (170 µl) was mixed with 340 µl ACN for deproteinization. After vigorous mixing, the samples were centrifuged for 3 min at 12,000 g and ambient temperature. The supernatant (400 µl) was mixed with eluent A–internal standard mix (800 µl) to transfer one part (400 µl) into a screw-capped glass HPLC sample vial for analysis in duplicate by LC-ESI MS/MS as described earlier. The remaining volume of the diluted supernatant was stored in a freezer at –20 °C for potential replication.
Standards
A tropane alkaloid cocktail containing all seven analytes in equal concentrations was produced for simultaneous analysis (1 µg/ml in 80:20 mix for atropine, cocaine, homatropine, ipratropium, littorine, N-butylscopolamine, and scopolamine). The highest-concentration standard in plasma was obtained by adding 30 µl of the cocktail to 270 µl human EDTA plasma (standard A, 100 ng/ml). An additional 11 calibration standards were generated by serial dilution (1:2) of each standard with 200 µl of blank plasma, resulting in concentrations ranging from 50 ng/ml (standard B) to 0.049 ng/ml (standard L). Plasma standards (170 µl) were immediately precipitated after spiking and were prepared for storage and measurement as described earlier. Two seven-point linear calibration curves of the peak area ratio of the analyte to the internal standard were used for quantification. Optimum linear regressions were obtained when the entire concentration range was subdivided into two segments: standard B to standard G (50–1.563 ng/ml) and standard F to standard L (3.125–0.049 ng/ml), each covering a calibration range of nearly 2 orders of magnitude.
If quantification of only a single analyte was necessary instead of a multicomponent analysis, calibration standards were prepared by spiking plasma (270 µl) with a tropane alkaloid solution (30 µl) containing the relevant analyte exclusively (1 µg/ml).
For stability studies in diluted human serum and rabbit serum, the preparation procedure for standards was modified accordingly in terms of matrix composition and dilution factors to cover the alkaloid concentrations applied (1 µg/ml), as described later.
Characteristics of LC-ESI MS/MS performance
Linear range, lower limit of quantification and detection, repeatability, and accuracy
Human EDTA plasma standards were prepared (n = 5) and analyzed as described earlier and additionally included lower-concentration standards (down to 0.003 ng/ml) to determine the lower limit of detection. Means and standard deviations of concentrations were calculated from the peak area ratio of the analyte to the internal standard to determine the linear range, the lower limit of quantification (LLOQ), precision (coefficient of variation, CV), and accuracy.
Ruggedness
The ruggedness of the method, describing the influence of slightly varying parameters during the analysis, was characterized by changing three selected parameters of the standard protocol (variant A) to slightly lower (variant B) and slightly higher (variant C) settings. The ionization spray voltage was changed from 4.8 to 4.7 and 4.9 kV, the HPLC flow rate was changed from 1.0 to 0.92 and 1.08 ml/min, and the gas spray pressure (heater gas/ion spray gas) was changed from 70/60 to 67/57 and 73/63 psi.
Two human EDTA plasma samples (500 µl each) were spiked with the seven-tropane alkaloid cocktail, resulting in concentrations of 10 and 0.83 ng/ml for each compound. Both samples were prepared following the principle of the standard protocol to produce two series of aliquots. Samples were analyzed using the LC-ESI MS/MS parameter conditions of variants A, B, and C in triplicate and quantified by a calibration curve measured under standard conditions (variant A).
Recovery and ion suppression
Recovery after precipitation of plasma and potential effects of ion suppression caused by matrix interferences during ionization of analytes were determined according to a procedure described earlier [27]. The principle behind the procedure includes the preparation of three calibration curves (sets 1–3) that differ in their composition of the matrix of the injection solution for LC-MS/MS analysis. Set 1 was prepared from spiked human plasma standards undergoing precipitation and dilution as described in the standard protocol. Set 2 consisted of standards that were prepared by spiking the decanted supernatant of precipitated blank plasma, thus avoiding loss of analyte due to incomplete recovery. Set 3 was produced in neat solvent, thus being influenced neither by any plasma matrix interferences nor by substance loss due to incomplete recovery. Comparison of the slopes of the three linear calibration curves allowed the calculation of both the average recovery (set 1/set 2) and ion suppression (set 2/set 3).
Injection volume
Human EDTA plasma was spiked with the seven-tropane alkaloid cocktail (12.5 ng/ml each) followed by precipitation and preparation according to the standard protocol described earlier. Increasing volumes of 20, 50, 75, 100, 130, 170, and 200 µl were analyzed by LC-ESI MS/MS in duplicate to determine the peak areas and the peak area ratios of the analyte to the internal standard.
Stability study of tropane alkaloids in PBS, human serum, and rabbit serum
Human serum and rabbit serum were diluted with PBS, resulting in 5% (v/v) solutions. Aliquots of 370 µl serum dilution as well as PBS alone were heated to 37 °C and spiked with tropane alkaloid solutions each containing only a single analyte (30 µl, 10 µg/ml in PBS for atropine, cocaine, homatropine, ipratropium, l-hyoscyamine, littorine, N-butylscopolamine, and scopolamine). Incubation mixtures (1 µg/ml) were kept at 37 °C under gentle shaking. Two samples of 30 µl each were collected immediately after spiking (t 0a, t 0b), followed by samples taken at eight additional distinct time points (t 1-t 8). On the basis of the results of pilot studies, these time points differed depending on the alkaloid tested, thus allowing us to monitor the optimum incubation period (Table 1). Only atropine was analyzed additionally at 20 time points in total, which was required for a comprehensive description of both the initial and the late phase of incubation (120 min).
Whereas incubations were carried out with single substrates, quantification was performed for all compounds simultaneously after pooling the prepared samples of each distinct time point (t 0a-t 8) to reduce the number of runs for analysis (Table 1). The sample preparation procedure was as follows. After precipitation with twice the volume of ACN (60 µl), 60 µl of the individual supernatants of all seven tropane alkaloids derived from one distinct time point were pooled (7 × 60 µl = 420 µl). Subsequently, 150 µl of the combined supernatants was further diluted with eluent A–internal standard mix and 80:20 mix. Analyses were performed in duplicate by LC-ESI MS/MS to quantify alkaloids simultaneously by an external calibration curve generated accordingly from standards prepared in a human serum dilution (5% v/v). Quantification of atropine (racemic d-hyoscyamine/l-hyoscyamine) was performed separately because differentiation from its single enantiomer l-hyoscyamine present in the sample pool was not possible.
Results and discussion
Tropane alkaloids possess eponymous chemical structures representing esters of a tropane derivative (azabicyclooctane moiety) and an organic acid, e.g., tropic acid in atropine/l-hyoscyamine (Fig. 1a), ipratropium (Fig. 1e), scopolamine (Fig. 1h), and N-butylscopolamine (Fig. 1g); mandelic acid in homatropine (Fig. 1b); 3-phenyllactic acid in littorine (Fig. 1f); and benzoic acid in cocaine (Fig. 1c). Owing to the related chemical structures, tropane alkaloids exhibit physiological activity similar to that of competitive antagonists of muscarinic receptors, which vary in their anticholinergic potency and pharmacological and toxicological properties [3, 4, 28]. We developed a LC-ESI MS/MS method as a generic procedure that enables quantitative measurement of such structural analogs, which comprise the most prominent tropane alkaloid drugs. Reliability was demonstrated by stability studies in rabbit serum and human serum allowing us to monitor time-dependent degradation.
MS characterization of tropane alkaloids
First, we characterized the MS behavior of the tropane alkaloids during collision-induced dissociation of the precursor ion after ESI in infusion experiments. Figure 1 shows the fragment spectrum, a list of the most intense signals, and the corresponding chemical structure of each alkaloid, indicating the cleavage sites of major fragments by dotted lines. Cleavage sites were deduced and assigned in analogy to the fragments of atropine [20, 21, 24] and scopolamine [29] described recently.
All alkaloids were detected as protonated singly charged molecular ions, with the exception of ipratropium (Fig. 1e) and N-butylscopolamine (Fig. 1g). The latter compounds possess a quaternary positively charged nitrogen atom, making the molecule detectable without proton addition. The fragmentation spectra were of different complexity but revealed similar cleavage sites for the alkaloids. In many cases the most abundant product ion signal was derived from the cleavage of the C-O bond connecting the tropane moiety with the acid residue, e.g., cleavage site b in atropine (Fig. 1a), site b in cocaine (Fig. 1c), and site c in scopolamine (Fig. 1h). Tropane structures possessing quaternary alkylated nitrogen (ipratropium, N-butylscopolamine) or MeO–CO– substitution (cocaine, cocaine-d 3) showed additional signals indicating the loss of these substituents. Cocaine-d 3 (m/z [M+H]+ 307.3; Fig. 1d) exhibited the same cleavage sites as cocaine (m/z [M+H]+ 304.1; Fig. 1c). Fragment ions containing the CD3 group bound to the nitrogen of the tropane moiety resulted in a ∆m/z of 3 Th when compared with those from unlabeled cocaine. As referred to Chen et al. [20], the signals at m/z 93.1 detected for atropine (Fig. 1a, signal c), homatropine (Fig. 1b, signal d), ipratropium (Fig. 1e, signal e), and littorine (Fig. 1f, signal d) are due to the loss of CH3–NH2 from the precursor ion at m/z 124 representing the cleaved tropane moiety. Accordingly, the signal at m/z 110.1 detected for scopolamine (Fig. 1h, signal e) and N-butylscopolamine (Fig. 1g, signal g) may be assigned to the corresponding oxidized ions derived from the cleaved scopine moiety.
In general, transitions from the (protonated) molecular precursor to the most abundant product ion of each alkaloid were elaborated by the automatic tuning mode of the mass spectrometer and used for quantification in the MRM mode as noted in Fig. 2.

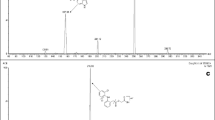
Simultaneous liquid chromatography (LC)–ESI MS/MS analysis of tropane alkaloids in plasma. a Total ion chromatogram of all seven alkaloids and cocaine-d 3 as an internal standard. Chromatography was performed in the gradient mode at 30 °C as indicated by the dashed line. Atlantis T3 C18 column, 5 µm, 150 mm × 4.6-mm inner diameter; eluent A 0.1% (v/v) FA, eluent B 80:20 (v/v) ACN/0.1% (v/v) FA; flow rate 1 ml/min; sample volume 100 µl. Mass-spectrometric detection was done in the positive multiple reaction monitoring mode: ionization spray voltage 4,800 V; curtain gas pressure 25 psi; heater gas pressure 70 psi; turbo ion spray gas pressure 60 psi; gas temperature 700 °C; entrance potential 10 V; dwell time 50 ms; collision gas nitrogen. b LC-ESI MS/MS of scopolamine eluted as peak 1, collision energy (CE) 29 V, declustering potential (DP) 61 V, collision cell exit potential (CXP) 8 V. c LC-ESI MS/MS of homatropine eluted as peak 1, CE 33 V, DP 41 V, CXP 6 V. d LC-ESI MS/MS of atropine eluted as peak 2, CE 35 V, DP 76 V, CXP 6 V. e LC-ESI MS/MS of littorine eluted as peak 3, CE 35 V, DP 81 V, CXP 6 V. f LC-ESI MS/MS of ipratropium eluted as peak 3, CE 45 V, DP 111 V, CXP 6 V. g LC-ESI MS/MS of cocaine (CE 29 V, DP 61 V, CXP 10 V) and cocaine-d 3 (CE 29 V, DP 61 V, CXP 12 V) eluted as peak 4. h LC-ESI MS/MS of N-butylscopolamine eluted as peak 5, CE 39 V, DP 71 V, CXP 8 V
Multicomponent analysis by LC-ESI MS/MS
Simple protein precipitation by the addition of twice the volume of ACN was used as a cheap, rapid, and effective plasma and serum sample preparation procedure with good recoveries for all tropane alkaloids tested (88–94%). Nevertheless, the analyte containing supernatant also comprised hydrophilic (e.g., salts, amino acids, carbohydrates) and more or less hydrophobic (e.g., peptides, remaining proteins, fatty acids, and phospholipids) matrix components. These compounds may affect the ionization process of coincidentally ionized analytes, thereby causing ion suppression and loss of intensity, which may negatively affect quantitative analysis. Therefore, LC separation was performed in a binary gradient mode to avoid or even minimize coelution of alkaloids and potential interferences. The influence of matrix ingredients on the ionization process was investigated systematically as described in “Recovery and ion suppression.” Figure 2a shows a typical total ion chromatogram obtained in the MRM mode of all seven alkaloids and the internal standard, also illustrating the applied gradient of eluent B containing ACN as an organic modifier. Analytes were eluted within a 4-min period. To realize efficient retention times and resolution of quite polar tropane alkaloids, a modern polar C18 stationary phase was used (Atlantis T3); this is highly superior to conventional reversed-phase material (results not shown). The excellent performance of the MRM mode allowed the highest selectivity in analyte detection even when alkaloids were coeluted, as occurred for peak 1 (scopolamine and homatropine, Fig. 2b, c), peak 3 (littorine and ipratropium, Fig. 2e, f), and peak 4 (cocaine and cocaine-d 3, Fig. 2g). As depicted in Fig. 1a and f, atropine (l-hyoscyamine) and its positional isomer littorine exhibit identical precursor (m/z 290.2) and product (m/z 124.1) ions, preventing MS differentiation. Therefore, the LC gradient method presented was developed to allow elution of both compounds with baseline separation as demonstrated in Fig. 2d and e, thus enabling selective detection and quantification.
Characteristics of LC-ESI MS/MS performance
We validated the LC-ESI MS/MS procedure to determine its reliability for complex matrices such as plasma. Calibration curves were obtained using the peak area ratio of the analyte to the internal standard. As no differences between the characteristics of l-hyoscyamine and atropine were observed, the results for both compounds are presented synonymously in the following (Table 2).
Linear range
The calibration curves (100-µl injection volume) for all seven alkaloids in plasma had excellent linearity (r 2 ≥ 0.999) when the standard concentration range was divided into two segments both covering nearly 2 orders of magnitude: 1.563–50 ng/ml (standard G to standard B; 17.4–555 pg on column) and 0.049–3.125 ng/ml (standard L to standard F; 0.54–34.7 pg on column). Concentrations of 100 ng/ml and greater were outside the linearity, presumably owing to detector overload.
This plasma concentration range is suitable for typical pharmacokinetic or toxicological analysis, e.g., about 20 µg atropine/ml after accidental overdosing (thus requiring further dilution of prepared samples) [4, 7, 21], 1–10 ng atropine/ml after usage of atropine autoinjectors for the treatment of organophosphorus poisoning [30, 31], 0.1–0.4 ng scopolamine/ml after transdermal dosage by a means of a patch or ocular administration [32], 1–1,000 ng ipratropium/ml in animal pharmacokinetic studies [33], 5 ng N-butylscopolamine/ml after oral intake [34], and 200–400 ng cocaine/ml after oral or intranasal drug abuse [35, 36]. Apart from these expected values, the LC-ESI MS/MS procedure was ideally suitable for the stability studies in human serum and rabbit serum reported here.
Lower limit of quantification
As listed in Table 2, the LLOQ for most tropane alkaloids was found for standard L (0.049 ng/ml plasma), characterized by sufficient precision (CVrepeatability≤13%) and accuracy (88–117%). The highest LLOQ was found for N-butylscopolamine (0.8 ng/ml) and was caused by unidentified matrix interferences from plasma. However, all limits of quantification were appropriate for the stability studies performed.
Lower limit of detection
With the exception of N-butylscopolamine, the chromatogram of which showed coeluted matrix interferences not identical with the tropane alkaloid itself, the lower limit of detection was found at concentrations as low as 0.004 and 0.01 ng/ml (Table 2).
Repeatability (intraday precision)
The CVrepeatability values for all standards (standard B to standard L) were typically below 7% in the middle region of the calibration curve for each alkaloid (n = 5). Close to the LLOQ, the CV values were found to not exceed 13%, thus underlining the high quality of the assay (Table 2).
Reproducibility (interday precision)
The CVreproducibility values determined from ten replicates measured during an 8-week period were below 10% in the middle region of the calibration curve. Corresponding accuracies were also of satisfactory quality in the range between 85 and 115% (results not shown).
Accuracy
As depicted in Table 2, the accuracies for all the analytes were between 87 and 117% over the entire concentration range, typically being 95% or more in the middle region, thus meeting the international requirements for accurate measurements [37]. Standards of lower concentration than the LLOQ exceeded the limitations (20% or more) and were therefore not considered for calibration.
Ruggedness
Ruggedness characterizes the impact of sometimes unavoidable changes of the system parameters that potentially influence analysis quality. Ionization spray voltage, HPLC flow rate, and gas spray pressure were varied as essential LC-ESI MS/MS parameters to slightly lower and higher values to determine the effect on precision, accuracy, and retention time. The ionization spray voltage was changed by ±0.1 kV (±2.1%) from the standard setting, which, in principle, influences molecule stability and ionization grade, thus affecting signal intensity. The HPLC flow rate was varied by ±0.08 ml/min (±8%), exerting a potential impact on separation performance (retention times, resolution). The gas spray settings were changed by ±3 psi (±5.0%), which generally affects the efficacy of the ionization process (desolvation, spray homogeneity). In common, none of these altered parameter settings caused significant changes in analyte quantification with respect to precision (CV mostly much below ±10%) and accuracy (100 ± 10%), as summarized in Table 3. The slightest impact was observed for cocaine (CV ±4%; accuracy 97–103%), which was due to the use of coeluted cocaine-d 3 as an internal standard undergoing absolutely identical changes. The retention times of all alkaloids varied within a range of ±0.2 min, demonstrating good reproducibility helpful for analyte assignment based on chromatographic retention. Therefore, the LC-ESI MS/MS procedure was proven to be very applicable for reliable analysis of plasma samples despite the use of only one common internal standard.
Recovery and ion suppression
The average recovery after plasma precipitation and ion suppression during ESI for each alkaloid was determined according to a procedure described recently [27]. The results obtained for atropine are illustrated in Fig. 3a and reveal high recovery (93 ± 3%) and nearly no ion suppression (97 ± 3% remaining intensity). Quite similar values (recovery/suppression) were calculated for each alkaloid: cocaine (94%/96%), homatropine (86%/96%), ipratropium (90%/96%), l-hyoscyamine (93%/97%), littorine (93%/99%), N-butylscopolamine (92%/99%), and scopolamine (88%/90%). These results indicate that plasma precipitation was an appropriate sample preparation procedure for each analyte and that ion suppression had a negligible influence.

a Calibration curves generated from atropine standards applied in different solvents to determine the recovery after plasma precipitation (93 ± 3%) and ion suppression caused by plasma matrix components (97 ± 3% remaining intensity). Black circles supernatant after precipitation of spiked plasma (set 1), gray circles spiked supernatant after plasma precipitation (set 2), triangles neat standard solution (set 3). The symbols represent the mean and the standard deviation of duplicate LC-ESI MS/MS analysis. Similar results of nearly quantitative recovery and negligible ion suppression were obtained for all other tropane alkaloids (results not shown). b Effect of increasing sample injection volumes on analyte peak area and corresponding peak area ratios (analyte to internal standard) exemplarily shown for atropine. Similar results of excellent linearity (injection volume vs peak area) and constant peak area ratios were obtained for all other tropane alkaloids (results not shown). The symbols represent the mean and the standard deviation of duplicate LC-ESI MS/MS analysis
Injection volume
In principle, accessible plasma sample volumes may depend on the study performed and can thus vary from a few milliliters for humans to some tens of microliters for small laboratory animals. As a consequence, the injection volumes available for LC-ESI MS/MS analysis may vary accordingly when an adapted sample preparation procedure is followed. Therefore, we investigated whether our method is also suitable for different injection volumes. Owing to the sample loop used (200 µl), increasing volumes (20–200 µl) of a prepared plasma sample were analyzed in duplicate to determine the corresponding analyte peak areas and peak area ratios (analyte to internal standard). Figure 3b demonstrates—exemplarily for atropine—that the resulting peak areas were of good linearity (r 2 = 0.997) and the analyte to internal standard ratios were constant (±2%) over the entire range of the injection volume. Furthermore, peak symmetries, retention times, and chromatographic resolution did not deteriorate. Similar data were obtained for all other tropane alkaloids tested (results not shown). These results underline the reliability of smaller and larger injection volumes for quantitative analysis, thus making the assay flexible and robust. We decided to use a 100-µl injection volume to reduce the amount of plasma sample needed and to reduce the risk of column or detector overload when analyzing unknown highly concentrated samples.
Exemplarily, we applied this quantitative multicomponent procedure to investigate the time-dependent concentration decrease of tropane alkaloids in rabbit serum.
Stability of tropane alkaloids in human serum and rabbit serum
Serum from rabbits is known to contain a polymorphic isoenzyme system of carboxylesterases (EC 3.1.1.1) that catalyze ester cleavage of some tropane alkaloids [38]. Atropinesterase (EC 3.1.1.10, also known as tropinesterase) cleaves tropic acid from, e.g., atropine and scopolamine, thus liberating tropine and scopine, respectively [39–41]. Atropinesterase has been detected in plants and in the serum and liver of rabbits, thus making rabbits resistant against the toxic effects of ingested berries and leaves of Atropa belladonna [39, 42]. In contrast, no corresponding activity was found in any other mammal, e.g., mouse, rat, guinea pig, goat, dog, rhesus monkey, and human [41, 43–45]. Although great effort has been made to characterize this enzyme phenomenon [40, 41, 43–46], no method has been described before that allows simultaneous analysis of tropane alkaloid degradation to evaluate the status of atropinesterase. Therefore, we decided to apply our multicomponent LC-ESI MS/MS procedure to monitor the degradation kinetics of tropane alkaloids in serum matrix.
Pilot studies using undiluted rabbit serum demonstrated very fast substrate degradation within a few seconds, thus preventing accurate and precise time-resolved measurement. Therefore, rabbit serum was diluted (5% v/v final concentration) to reduce the enzymatic activity of the incubation mixture. In initial studies, human serum was used for dilution to keep the maximum realistic complexity of the sample matrix, allowing us to document the reliability of the LC-ESI MS/MS procedure for serum analysis (results not shown). No impact on the analytical performance was observed when serum was used instead of plasma.
However, this procedure prevented clear evaluation of enzymatic activity of rabbit serum enzymes owing to the influence of human serum components potentially causing inhibition or additional degradation. Therefore, rabbit serum was diluted in PBS to exclude this impact. Furthermore, tropane alkaloids were also incubated as references under identical conditions in (1) PBS alone to elucidate nonenzymatic hydrolysis and (2) in diluted human serum to characterize non-rabbit enzyme activity.
None of the tropane alkaloids tested was degraded in PBS incubation buffer, thus excluding nonenzymatic hydrolysis (Fig. 4a–h).

Stability of tropane alkaloids in diluted human serum and rabbit serum and in phosphate-buffered saline (PBS). Human serum and rabbit serum were diluted in PBS to result in 5% (v/v) solutions at pH 7.6. Tropane alkaloids were added to diluted serum and PBS as single substrates (all 1 µg/ml) followed by incubation at 37 °C under gentle shaking. The symbols represent the mean and the standard deviation of duplicate LC-ESI MS/MS analysis. Circles PBS, squares human serum, triangles rabbit serum. a Stability of atropine (racemic d-hyoscyamine/l-hyoscyamine) within 16 min of incubation (rabbit serum τ 1/4 = 4.6 min); insert incubation of atropine in diluted rabbit serum monitored for 2 h. Within about 20 min atropine was degraded by 50% (enantioselective degradation of l-hyoscyamine). No further degradation appeared within the following incubation period. The concentration plateau was caused by remaining d-hyoscyamine. b Stability of l-hyoscyamine within 8 min of incubation (rabbit serum τ 1/2 = 2.6 min). c Stability of littorine within 90 min of incubation (rabbit serum τ 1/2 = 13.4 min). d Stability of l-scopolamine within 60 min of incubation (rabbit serum τ 1/2 = 3.9 min). e Stability of N-butylscopolamine within 160 min of incubation (no degradation). f Stability of ipratropium within 160 min of incubation (no degradation). g Stability of homatropine within 90 min of incubation (rabbit serum τ 1/2 = 15.1 min). h Stability of cocaine within 60 min of incubation (rabbit serum τ 1/2 = 32.0 min). In contrast to all other tropane alkaloids, significant degradation in human serum was observed (human serum τ 1/2 = 130 min).
Whereas l-hyoscyamine was degraded completely in rabbit serum within 9 min, exhibiting a period of half change (τ 1/2) of 2.6 min (Fig. 4b), atropine concentrations were only reduced to 50% of the initial concentrations, reaching a stable plateau after about 20 min (Fig. 4a). These findings indicate that (1) ester cleavage catalyzed by atropinesterase is an enantioselective process highly favoring the natural l enantiomer and (2) d-hyoscyamine acts as a competitive inhibitor of l-hyoscyamine degradation as is obvious from the longer periods of half change observed for l-hyoscyamine in the presence of its d enantiomer (τ 1/4 = 4.6 min). Our results are in accordance with the findings of Werner [41], who stated that enzymatic cleavage of l-hyoscyamine appears to be 80 times faster than that of its d enantiomer. Similar effects of enantioselectivity have also been shown for enzymes isolated from rabbit serum and liver homogenates also cleaving scopolamine, homatropine, and cocaine [41]. The degradations of littorine (Fig. 4c, τ 1/2 = 13.4 min) and l-scopolamine (Fig. 4d, τ 1/2 = 3.9 min) followed a continuous exponential decay, whereas the concentration–time profile of d-homatropine/l-homatropine appeared as a biphasic decrease (Fig. 4g). Within the first 15 min the concentration decreased rapidly to 50% (τ 1/2 = 15.1 min), but was followed by a much slower decrease indicating enantioselective ester cleavage. Cocaine, present as its natural l enantiomer not undergoing racemization, was degraded in rabbit serum and exhibited a period of half change of 32.0 min (Fig. 4h). This decrease was due to cocainesterase—an enzyme not identical to atropinesterase—cleaving benzoic acid from methyl ecgonine [38, 47].
In contrast to all the other alkaloids tested, cocaine was also degraded in human serum (Fig. 4h), catalyzed by butyrylcholinesterase (EC 3.1.1.8) [48].
In contrast to the above-mentioned tropane alkaloids possessing a tertiary amine in the tropane moiety, the quaternary amine derivatives N-butylscopolamine and ipratropium did not show any degradation in rabbit serum (Fig. 4e, f). This appears to be due to their doubly alkylated nitrogen atom preventing enzymatic degradation, presumably owing to either the permanent positive charge or steric hindrance.
Further analysis of enzymatic degradation with respect to elucidation of relevant enzymes and kinetics is beyond the scope of this application and is therefore not discussed in more detail. However, this application demonstrates the benefit of simultaneous quantification and was suitable for elucidation of the time-dependent concentration decrease as may also occur in pharmacokinetic studies.
Conclusions
We described the development of a quantitative analytical procedure to determine seven tropane alkaloids simultaneously by LC-ESI MS/MS. This validated procedure was shown to be reliable and very suitable for the quantification of the most prominent anticholinergic drugs which are of toxicological relevance in the case of, e.g., drug abuse, overdosing, and accidental intake by ingestion of berries and leaves from nightshade plants. To document the valuable properties of this multicomponent method, the stability of relevant tropane alkaloids against serum enzymes was investigated. Whereas no significant degradation was observed in human serum (except esteratic cleavage of cocaine by butyrylcholinesterase), rabbit serum enzymes degraded all tropane alkaloids possessing a tertiary amine in the tropane moiety. With respect to earlier studies elucidating esteratic cleavage of such alkaloids, enantioselective activity of atropinesterase and cocainesterase was observed, which is a unique mammalian phenomenon found exclusively in rabbits. The quantitative method presented will thus be valuable for pharmacokinetic and toxicokinetic studies as well as for forensic samples, stability studies, and enzymatic studies.
References
Kutchan TM (1998) In: Cordell GA (ed) The alkaloids, chemistry and biology, vol 50. Academic, San Diego, pp 295–303
Schmeller T, Sporer F, Sauerwein M, Wink M (1995) Pharmazie 50:493–495
Reynolds JEF (ed) (1996) Martindale the extra pharmacopoeia, 31st edn. Royal Pharmaceutical Society, London
Heath AJW, Meredith T (1992) In: Ballantyne B, Marrs TC, Aldrige WN (eds) Clinical and experimental toxicology of organophosphates and carbamates, 1st edn. Butterworth-Heinemann, Oxford, pp 543–554
Virtanen R, Kanto J, Iisalo E (1980) Acta Pharmacol Toxicol 47:208–212
Wurzburger RJ, Miller RL, Boxenbaum HG, Spector S (1977) J Pharmacol Exp Ther 203:435–441
Bogan R, Zimmermann T, Zilker T, Eyer F, Thiermann H (2009) Clin Toxicol 47:602–604
Thiermann H, Radtke M, Spöhrer U, Klimmek R, Eyer P (1996) Arch Toxicol 70:293–299
Tahara SI, Okayama A, Kitada Y, Watanabe T, Nakazawa H, Kakehi K, Hisamatu Y (1999) J Chromatogr A 848:465–471
Bo T, Li KA, Liu H (2003) J Pharm Biomed Anal 31:885–891
Wintersteiger R, Gamse G, Pacha W (1982) Fresenius' Z Anal Chem 312:455–461
Kaplan MM, Register DC, Bierman AH, Risacher RL (1974) Clin Toxicol 7:509–512
Patterson S, O'Hagan D (2002) Phytochemistry 61:323–329
Kehe CR, Lasseter KC, Miller NC, Wick KA, Shambleen EC, Ekholm BP, Sandahl JH, Chang SF, Goldlust MB, Kvam DC, Harrison LI (1992) Ther Drug Monit 14:132–134
Kirchhoff C, Bitar Y, Ebel S, Holzgrabe U (2004) J Chromatogr A 1046:115–120
Büch U, Isenberg E, Büch HP (1994) Methods Find Exp Clin Pharmacol 16:361–365
Ceyhan T, Kartal M, Altun ML, Tülemis F, Cevheroglu S (2001) J Pharm Biomed Anal 25:399–406
Takahashi M, Nagashima M, Shigeoka S, Nishijima M, Kamata K (1997) J Chromatogr A 775:137–141
Lau OW, Mok CS (1997) J Chromatogr A 706:270–276
Chen H, Chen Y, Du P, Han F, Wang H, Zhang H (2006) J Pharm Biomed Anal 40:142–150
Boermans PAMM, Go HS, Wessels AMA, Uges DRA (2006) Ther Drug Monit 28:295–298
Kintz P, Villian M, Barguil Y, Charlot JY, Cirimele V (2006) J Anal Toxicol 30:454–457
Johansen SS, Bhatia HM (2007) J Chromatogr B 852:338–344
Beyer J, Peters FT, Kraemer T, Maurer HH (2007) J Mass Spectrum 42:621–633
Björnstad K, Beck O, Helander A (2009) J Chromatogr B 877:1162–1168
Steenkamp PA, Harding NM, van Heerden FR, van Wyk BE (2004) Forensic Sci Int 145:31–39
John H (2005) Anal Bioanal Chem 38151–53
Brown JH, Taylor P (1996) In: Hardman JG, Limbird LE (eds) The pharmacological basis of therapeutics, 9th edn. McGraw-Hill, New York, pp 141–160
Chen H, Chen Y, Wang H, Du P, Han F, Zhang H (2005) Talanta 67:984–991
Friedl KE, Hannan CJ, Schadler PW, Jacob WH (1989) J Pharm Sci 78:728–731
Kamimori GH, Smallridge RC, Redmond DP, Belenky GL, Fein HG (1990) Eur J Clin Pharmacol 39:395–397
Renner DU, Oertel R, Kirch W (2005) Ther Drug Monit 27:655–665
Leusch A, Eichhorn B, Müller G, Rominger KL (2001) Biopharm Drug Dispos 22:199–212
Tytgat GN (2007) Drugs 67:1343–1357
Van Dyke C, Jatlow P, Ungerer J, Barash PG, Byck R (1978) Science 200:211–213
Javaid JI, Fishman MW, Schuster CR, Dekirmenjian H, Davis JM (1978) Science 202:227–228
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Veterinary Medicine (2001) Guidance for industry: bioanalytical method validation
Van Zutphen LFM (1974) Biochem Genetics 12:309–326
Moog P, Krisch K (1974) Hoppe Seylers Z Physiol Chem 355:529–542
Linn JM, Liebenberg SP (1979) Lab Anim Sci 29:335–337
Werner G (1961) Planta Med 4:293–316
Schroff CD (1852) Z KK Gesell Arzte Wien 3:211–242
Harrison PK, Tattersall JEH, Gosden E (2006) Naunyn Schmiedebergs Arch Pharmacol 373:230–236
Fox RR, Tucker FS (1984) Lab Anim Sci 34:381–382
Tucker FS, Beattie RJ (1983) Lab Anim Sci 33:268–269
Margolis F, Feigelson P (1963) J Biol Chem 238:2620–2627
Stewart DJ, Inaba T, Tang BK, Kalow W (1977) Life Sci 20:1557–1564
Duysen EG, Bartels CF, Lockridge O (2002) J Pharmacol Exp Ther 302:751–758
Acknowledgements
We thank Koichiro Shimomura (School of Life Sciences, Toyo University, Gunma, Japan) for providing the synthetic littorine standard and Steffen Krüger and Johann Baur (Bundeswehr Institute of Pharmacology and Toxicology) for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
John, H., Binder, T., Höchstetter, H. et al. LC-ESI MS/MS quantification of atropine and six other antimuscarinic tropane alkaloids in plasma. Anal Bioanal Chem 396, 751–763 (2010). https://doi.org/10.1007/s00216-009-3209-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-3209-7