
Abstract
A method of standoff detection based on the observation of laser-induced fluorescence–amplified spontaneous emission (LIF-ASE) is described. LIF-ASE generates uniaxial intensity distributions of the observed fluorescence with the majority of intensity propagating along the excitation axis in both the forward and backward directions. The detection of bulk vapor at significant standoff distances is readily achieved. This method was used to detect NO directly and as a photoproduct after 213-nm excitation of 2-, 3-, and 4-nitrotoluene. The NO LIF-ASE spectra were studied as a function of buffer gas. These studies showed that the emission from different vibrational states was dependent upon the buffer gas used, suggesting that the populations of vibrational states were influenced by the environment. A similar sensitivity of the vibrational populations was observed when the different nitroaromatic precursors were used in nitrogen buffer gas. Such sensitivity to environmental influences can be used to distinguish among the different nitroaromatic precursors and facilitate the identification of the bulk vapor of these analytes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Methods useful for trace materials detection have been subjected to intense research over the last decade and significant progress has been made. Analytical methodologies to detect explosive residues are classified as either contact or standoff methods depending upon the method used. Contact analysis requires sample collection for off-line analysis or direct analysis of the sample using a portable field device [1, 2]. Standoff detection allows monitoring to be achieved remotely, at discreet distances, and generally uses pulsed lasers to detect and identify trace materials. Laser-based detection of explosives has been the subject of a recent review [3].
Primary explosives (including nitroaromatics and nitramines) absorb in both the infrared (IR) and ultraviolet (UV) spectral region, allowing their analysis via vibrational and electronic spectroscopies. Trace detection of explosives has been demonstrated using Raman [4, 5], IR absorption [6, 7], IR and UV cavity-ring-down spectroscopies [8, 9], coherent anti-Stokes Raman spectroscopy (CARS) [10], and Terahertz [11, 12] methods. Photofragmentation followed by laser-induced fluorescence (PF-LIF) detection schemes use laser pulses to fragment the analyte and subsequently identify at least one of the fragments via laser-induced fluorescence [13–15]. Laser-induced breakdown spectroscopy (LIBS) relies on plasma emission. LIBS has been used successfully to detect a number of primary explosives based on chemometric analysis of the plasma emissions observed [16]. Deconvolution of the analyte signal from that of air and/or the substrate remains a daunting challenge [17].
The majority of optical standoff detection methods focus on trace detection, where the requirement of hitting the solid or liquid target with the laser pulse severely limits the time needed to interrogate a large object or area. Although a number of all-optical standoff detection schemes have been demonstrated for trace explosives detection, they all suffer from the shortcomings imposed by the need to raster scan areas or objects under investigation. In addition, interference from the coatings (i.e., paint, waxes, etc.) or innocuous contaminants on the surface can distort the observed spectra. Bulk detection of vapor clouds offers the opportunity to overcome these limitations. The laser pulse would not strike a surface or substrate, but would interact directly with the analyte vapor as it emanates from samples. Successful demonstration of bulk vapor detection at standoff distances would provide new analytical capabilities for surveillance. The intrinsically low vapor pressure of most primary explosives is a significant challenge to this approach. New strategies must be identified to increase the intensity of the backscattered signal.
One advantage of using all-optical detection methods is the potential to employ coherent laser-stimulated processes to enhance the intensity and directionality of the signal. The detection of explosives using coherent laser methods has already been accomplished with CARS spectroscopy [10]. Because all energetic materials absorb in the UV, spectroscopic signals can originate from UV-generated excited states or photofragments of the material of interest. Laser-induced fluorescence–amplified spontaneous emission (LIF-ASE) is a spectroscopic technique that is well-suited for the standoff detection of chemical vapors [18–20]. This technique uses high-peak-power laser pulses to produce cylindrical interaction volumes of excited states within the vapor as the light pulse travels through the sample. Photons emitted from these excited states in directions perpendicular to the cylindrical axis leave the interaction volume with little opportunity to encounter additional excited states. These photons are observed as spontaneous emission. In contrast, photons emitted along the cylindrical axis have a significant probability of interacting with additional excited states and thereby induce stimulated emission. Thus, amplified spontaneous emission (ASE) can be observed propagating in both directions along the cylindrical axis of the interaction volume. Forward-propagating emission follows the initial laser pulse away from its source while the backward-propagating emission returns to the source as a well-defined pulse. Because the ASE signal is intrinsically directional, standoff detection is feasible at distances ranging from a few centimeters to more than a kilometer.
The stimulated nature of coherent spectroscopy has advantages over conventional absorption, emission, or Raman spectroscopy by virtue of photon amplification afforded by its coherent nature. It is anticipated that both a critical laser intensity and analyte concentration will be required to breach the threshold before significant ASE occurs. Nevertheless, the signal amplification associated with these processes should offer dramatic rewards in detection limits when compared with the detection of the spontaneous processes under otherwise identical conditions.
Of the optical detection methods described above, LIF offers a sensitive approach to standoff detection and is suitable for detecting compounds that absorb in the UV spectral region, including aromatic hydrocarbons and fluorescent decomposition products (e.g., nitric oxide) from non-fluorescent analytes. The detection of nitrated-toluene and -benzene vapors via fluorescent NO and NO2 photofragments has been demonstrated in the ppb to ppm range [13–15]. This manuscript describes the detection of backscattered LIF-ASE from vapor phase NO photofragments using a standoff technique developed in our laboratory.
Experimental
Materials
Samples of 2-nitrotoluene (2NT; 99+%), 3-nitrotoluene (3NT; 99%), and 4-nitrotoluene (4NT; 99%) were obtained from Aldrich and used as received. Argon (Ar) and helium (He) buffer gasses were purchased from Puritan Bennett as 99.995% purity while nitrogen (N2) buffer gas was obtained from a liquid N2 boil-off gas delivery system. Nitric oxide (NO) was produced in situ by adding nitric acid dropwise onto copper metal under a stream of N2 gas [21]. The NO samples were passed through a cold trap utilizing a LN2/methanol slurry before being diluted with additional buffer gas. The samples were then allowed to flow through the sample tube, as described below, for spectral measurements. Based on the respective flows of generated NO and buffer gas, the concentration of NO was estimated to be about 50 ppm within the sample tube.
LIF-ASE measurements
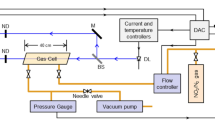
The LIF-ASE spectrometer is shown schematically in Fig. 1. A Q-switched Nd:YAG laser (Continuum Surelight II) was used to produce ca. 8-ns pulse width 1,064-nm pulses at a 10-Hz repetition rate. The fundamental was frequency-doubled and then quadrupled using the commercial KDP crystal set. Residual 532-nm light was removed using a 10-cm cell containing aqueous CoSO4. The fifth harmonic at 213 nm was generated by mixing the 1,064-nm fundamental and 266-nm fourth harmonic in an angle-tuned BBO crystal. Three dichroic harmonic separators (CVI) were used to separate the residual fundamental and fourth harmonic from the 213-nm pulse and, in turn, direct the 213-nm pulse through the LIF-ASE apparatus. The beam diameter was reduced to 2-mm using an uncoated quartz telescope for subsequent excitation of the samples. These excitation pulses were launched down the center of the sample tube using the final 213-nm harmonic separating beam splitter. After passing through the sample tube the residual 213-nm radiation was collected in a beam dump located 2 m past the sample compartment. Backscattered LIF-ASE generated within the sample tube was wavelength-selected on the return pass through the harmonic separator; wavelengths shorter than ca. 225 nm being rejected while longer wavelengths were transmitted. The transmitted LIF-ASE passed through an adjustable iris and was coupled into a quartz fiber optic using an uncoated quartz lens. The distal end of the optical fiber was attached to the input arm of an Acton SpectraPro 300i spectrograph. The typical distance between the collection lens and the center of the sample tube was ca 1.2 m but measurements at distances of 15 m using this collection scheme have been accomplished with comparable results. The spectrograph was equipped with a 300 nm/300 groves mm−1 grating and the output detected using a 1,064 × 256 CCD (Roper Scientific). Typical exposure times of 25 s were used. This arrangement allowed ca 0.8 nm resolution spectra to be collected. Spectra were not corrected for the wavelength-dependent detector response or the transmittance of the optical components. The intensities were measured as counts recorded at the peak of the spectrally resolved vibronic bands.

Schematic of the LIF-ASE apparatus used to study vapor samples. (a) 213-nm harmonic separators, (b) uncoated quartz collimating telescope to reduce beam diameter by 4×, (c) windowless double-jacket sample vapor tube, (d) beam dump, (e) pcx focusing lens, (f) fiber optic coupler
Vapor samples of the nitroaromatics were produced by flowing buffer gas over samples of solids, or through filter papers wetted with liquid samples. The concentrations of vapor were adjusted by using supplemental flow of buffer gas mixed into the primary stream before entering the sample tube. Typical total buffer gas flow rates of 300 L/hr at atmospheric pressure were used in all cases. The sample tube was a 1″-diameter windowless tube 60 cm long with a hose connection to allow gas mixtures to be introduced into the center of the tubes length. Alternatively, neat solid or liquid samples were placed on the bottom of the sample tube into which buffer gas was flowed at ca. 20 ml/min. For 2NT and 3NT, the sample tube was allowed to reach steady-state concentrations at room temperature, while for 4NT the sample tube containing solid 4NT was heated to about 70 °C. Based on the flow rates and saturation vapor pressures of these nitroaromatics the analyte concentrations were estimated to be on the order of 100 ppm.
Results
The absorption spectrum of NO in N2 buffer gas is shown in Fig. 2. There is a clear progression of vibronic transitions from the X 2Π v″ = 0 ground state into several vibrational levels of the A 2Σ first excited state. Additional progressions into higher excited states are also observed. The Nd:YAG fifth harmonic (dashed line in Fig. 2) occurs at 213 nm and coincides with the A 2Σ–X 2Π (1,0) transition. Thus, absorption of 213-nm light populates v′ = 1 of the A 2Σ excited state.

LIF-ASE spectra observed for direct excitation of NO in N2 (black line), Ar (blue line), and He (red line) buffer gasses after 213-nm excitation (shown as vertical dashed line) at atmospheric pressure. The absorption spectrum of NO is included (gray line) for reference
The LIF-ASE spectrum of NO was obtained by passing 213-nm laser pulses (ca. 0.5 mJ pulse; 8-ns pulsewidth) through a flowing sample of NO. The observed emission spectrum in N2 buffer gas is shown in Fig. 2. This spectrum was not corrected for the wavelength-dependent detector response or the transmittance of the optical components including the harmonic separators used to direct the 213-nm excitation pulse and reject the return scattered light. Emissions near 225 nm and below will be largely rejected by the harmonic separator but wavelengths above 250 nm will be largely transmitted. Emissions observed between these two limits will show increasing attenuation as the wavelength decreases. The signal observed at 213 nm is dominated by scattered laser light which was not completely rejected by this separator. The observed emission spectrum clearly shows three vibrational progressions. Emissions originating from different vibrational states are indicated along the top of Fig. 2. The assignments, frequencies, and relative intensities of these transitions are collected in Table 1. The frequencies of these transitions correspond to those reported previously [22].
When Ar or He were used as buffer gasses, the relative intensities of the vibronic bands were altered relative to those observed in N2. These LIF-ASE spectra are also show in Fig. 2 and the relative emission intensities in Ar and He are included in Table 1.
LIF-ASE was not observed when 266-nm excitation (up to 40 mJ pulse energies) was used as the pump source to excite 2-nitrotoluene (2NT) vapor. It has been shown that NO can be produced from nitroaromatic precursors using laser pulses in the 210- to 270-nm range [23]. However, as indicated in Fig. 2, NO does not absorb 266-nm light and therefore no LIF is observed using the Nd:YAG fourth harmonic. In contrast, strong emission is observed when 213-nm pump pulses (0.5 mJ) are used to excite the same samples. The NO produced photochemically within a single 8-ns pulse is able to absorb the 213-nm light and subsequently emit it. The emission spectrum observed is readily identified as belonging to NO and is shown in Fig. 3. The relative intensities for each of the observed emissions are also included in Table 1.

LIF-ASE spectra observed after 213-nm excitation of vapors of NO, 2NT, and 3NT at room temperature and 4NT at 70 °C. In each case, the buffer gas was N2 at atmospheric pressure
Comparable spectra were obtained when 3-nitrotoluene and 4-nitrotoluene were used as the photochemical source of NO and these emission spectra are also included in Fig. 3 and the relative intensities are again recorded in Table 1. Both 2NT and 3NT were sufficiently volatile at room temperature (23 °C) to be observed. The sample tube was heated in the case of 4NT such that the emission intensity was observed to increase upon relatively minor heating and reached a maximum at about 70 °C.
The power dependence of 2NT emission intensity in N2 buffer gas was also studied. Three plots of the observed intensity for specific transitions versus excitation power are shown in Fig. 4. The slopes of these plots are 3.2 ± 0.5 for transition #11, 3.5 ± 0.3 for transition #3, and 3.6 ± 0.4 for transition #18.

Ln–ln plots of the LIF-ASE emission intensity observed upon 213-nm excitation of 2NT in N2 buffer gas versus the excitation intensity. Individual plots depict the intensities for transitions originating from different vibronic states within the spectrum, i.e., transition #11 (triangles slope 3.2 ± 0.5), transition #3 (circles slope 3.5 ± 0.3), and transition #18 (diamonds slope 3.6 ± 0.4)
Discussion
It is well known that NO is a product in the photofragmentation of nitroaromatics. PF-LIF has been used to detect TNT vapor and TNT adsorbed in soil and groundwater [15, 24, 25]. Nitrobenzenes, nitrotoluenes, and nitromethane have also been detected, in some cases at concentrations down to ppb [13, 26]. ASE offers promise for the detection of low-vapor-pressure materials, with signal enhancements as high as 104 over spontaneous fluorescence [18]. In addition to the signal enhancement, the directionality of the backscattered emission offers the possibility of LIF-ASE detection at significant standoff distances.
The resolution used to collect the spectra shown in Figs. 2 and 3 clearly show vibrationally resolved transitions. The rotational progressions within each vibronic band are not resolved. The frequencies of the NO vibronic bands shown in Figs. 2 and 3 match those previously reported [22]. The transition intensities are dependent on the bath gas used, although they have not been corrected for the wavelength-dependent response of the spectrograph, the CCD or the optical components used in apparatus.
The spectrally resolved LIF from the diatomic photofragment offers advantages over LIF from the parent molecule. First, nitrotoluenes do not fluoresce, but fragment, from the electronically excited state. In addition, the vibronic bands in the luminescence spectrum of polyatomics are significantly broader than those shown in Figs. 2 and 3. A well-resolved spectral fingerprint is useful in the positive identification of an analyte.
Three separate vibronic progressions can be identified in the LIF-ASE spectrum of NO and the nitrotoluenes. One of these progressions originates from A 2Σ v′ = 1, which is populated directly by a 213-nm excitation. A second progression is identified as originating from the A 2Σ v′ = 0 excited state, which is presumably populated by collisional deactivation within the excited-state lifetime of NO [27]. The third observed progression is readily identifiable as originating from the A 2Σ v′ = 2 excited state.
Comparisons among the NO LIF-ASE spectra obtained in different buffer gasses show that the vibrational populations are influenced by the nature of the buffer gas. As seen in Fig. 2, the emission intensities arising from the A 2Σ v′ = 1 and A 2Σ v′ = 0 states are of comparable intensities when N2 is used as buffer gas. In contrast, the spectra recorded in the monoatomic Ar or He buffer gasses show that transitions originating from the A 2Σ v′ = 1 state are significantly more intense than those originating from A 2Σ v′ = 0. Not surprisingly, these results indicate that the rate of vibrational energy transfer from NO to N2 buffer gas is faster than collisional deactivation by Ar or He. The dependence of collisional deactivation rate on buffer gas is well known. As described below, the sensitivity of the deactivation rates to environmental influences is important in the determination of the identity of the parent nitroaromatic species from which the NO fragment is produced.
The emission spectra obtained after 213-nm excitation of 2NT, 3NT, and 4NT vapors in N2 buffer gas at atmospheric pressure are shown in Fig. 3. The spectra observed for these nitroaromatics share the same vibronic progressions as those observed from the direct excitation of NO. However, comparisons among the spectra do show the populations of the vibrational levels within the A 2Σ excited state are dependent upon the precursor used. For example, as can be seen in Fig. 3, the transitions originating from the A 2Σ v′ = 1 state are more intense than those originating from A 2Σ v′ = 0 in the case of 3NT but the transitions originating from the A 2Σ v′ = 1 are more intense in both 2NT and 4NT. In addition, all of the NT isomers show differences from NO. Specifically, in NO, the A 2Σ → X 2Π (1,4) transition at 255.4 nm is more intense than the A 2Σ → X 2Π (0,3) transition at 259.1 nm. The relative intensities of these two vibronic peaks are reversed in the NTs. The absolute value of these intensity ratios will be dependent upon the relative sensitivity of the detector and the transmittance of the optical components at the individual wavelengths, which has not been corrected in the current report. Of importance to the current discussion is the fact that these ratios are indeed different and this difference will persist even when such corrections can be made. Similarly, a comparison of the intensities for transition originating from A 2Σ v′ = 2 leads to the conclusion that these intensities follow the ordering 2NT > 4NT ∼ 3NT. The fact that the spectral patterns change suggests that these patterns can be used to identify the precursors from which the NO originates.
The intensity differences observed may be indicative of different excited-state populations due to the isomer-dependent photofragmentation [23, 28, 29] although it is unlikely to be the single cause for the observed phenomenon. It seems unreasonable to suggest that isomer-dependent photofragmentation could result in a higher degree of vibrational relaxation after excitation of the photoproduct NO than is observed when NO is excited directly, as is observed here. Instead, it is possible that the environment surrounding the NO photoproduct modifies the rate of vibrational relaxation. The photoproduct environment will depend upon both the fragmentation mechanism and the structures of the fragments.
If such intensity patterns are to be useful as a tool to identify the precursors from which the NO is produced, their appearance should not be sensitive to the conditions of the experiment. For this reason, the excitation laser pulse power dependence on LIF-ASE spectra of 2NT was examined. Three plots of the observed intensity for specific transitions versus excitation power are shown in Fig. 4. In each case, the slope of the plots exceeds 3, indicating a highly non-linear dependence of emission intensity on the excitation intensity. In contrast, the power dependences of the LIF-ASE signals observed for direct excitation of NO are nearly linear (data not shown). The observation of slopes greater than 1 in these plots could have been predicted because the photochemical production of NO from nitroaromatic precursors and the subsequent excitation of the NO produced is a multiphoton in nature. The magnitude of the slopes observed will have a complex dependence upon the concentration profiles of the absorbers, their absorption cross-sections, the laser fluence, and saturation behavior. A complete analysis of their magnitudes is beyond the intended scope of this report. More germane to this discussion, at least from an analytical perspective, is that the power dependence for the appearance of each vibronic series is similar and therefore the shapes of the observed emission spectra do not change within the range of excitation energies used (i.e., between 1.0–0.15 mJ). Identification of specific nitrotoluenes via the characteristic vibronic fingerprint will not be affected when the laser pulse powers used are within this range. How the observed emission intensity changes as a function of concentration has yet to be investigated systematically. Based on preliminary studies with differing concentrations of analytes and observing the signal-to-noise recorded in the spectra shown in Figs. 2 and 3 suggest that standoff detection of NO at concentrations significantly lower than 10 ppm can be achieved. While the room-temperature vapor pressure of TNT is several orders of magnitude lower than 10 ppm, these results show that many analytes (including acetone and combustible fuels) as well as ICAO taggants [30] (2NT and 4NT) with vapor pressures falling in this range can be detected and identified. Strategies for increasing the vapor phase concentrations of low-vapor-pressure analytes using multiple laser pulses in well-defined sequences are currently being explored in our laboratory.
There are two important questions that remain to be addressed. First, why not correct these spectra for the wavelength-dependent response of the CCD or the optical transmittance of the components used in apparatus? And second, is ASE really observed or is the emission recorded simply spontaneous emission collected using a favorable geometry? The answers to these two questions are inextricably intertwined. Emission spectra of NO have been reported on an absolute scale such that correction of the experimental apparatus could be attempted based solely on the data presented within Fig. 2. All that would be required would be to insist that the shape of the observed emission spectrum correspond to that predicted based on the reported Franck–Condon factors for these transitions. Unfortunately, the reported Franck–Condon factors cannot account for spectra in which a significant contribution due to ASE exists. A review of the data in Table 1 shows that the relative emission intensities reported therein are systematically different even for transitions originating from the same excited state. Thus, the simple method for correcting these spectra is not valid because ASE contributes to the observed emission. Alternative methods of correcting the optical response are being explored.
Conclusion
A new all-optical standoff method of detecting low vapor pressure analytes has been described. The detection scheme involves the well-known photofragmentation-LIF of nitrotoluenes and offers several operational advantages as an analytical technique. The use of a Nd:YAG harmonic provides a more robust source for interrogation than a tunable solid-state or dye laser. In addition, design of field apparatus is simplified due to the fixed-frequency LIF-ASE spectra. Finally, the approach allows for the identification of specific isomers (3NT vs. 2NT and 4NT), as well as the ability to discern the NTs from NO. Further experiments are underway to identify and characterize the temporal characteristics of additional organic photofragments to enhance further the multimodal aspects and sensitivity of this standoff detection scheme for explosives with low vapor pressure.
References
Woodfin RL (2007) Trace chemical sensing of explosives. Wiley, Hoboken, NJ, p 363
Moore DS (2007) Sensing and Imaging 8:9–38
Yinon J (2007) Counterterrorist detection techniques of explosives. Elsevier, London
Sharma SK, Misra AK, Lucey PG, Angel SM, McKay CP (2006) Appl Spectrosc 60:871–876
Carter JC, Angel SM, Lawrence-Snyder M, Scaffidi J, Whipple RE, Reynolds JG (2005) Appl Spectrosc 59:769–775
Janni J, Gilbert BD, Field RW, Steinfeld JI (1997) Spectrochimica Acta Part A-Molecular and Biomolecular Spectroscopy 53:1375–1381
Riris H, Carlisle CB, McMillen DF, Cooper DE (1996) Appl Opt 35:4694–4704
Todd MW, Provencal RA, Owano TG, Paldus BA, Kachanov A, Vodopyanov KL, Hunter M, Coy SL, Steinfeld JI, Arnold JT (2002) Appl Phys B-Lasers Opt 75:367–376
Ramos C, Dagdigian PJ (2007) Appl Opt 46:620–627
Portnov A, Rosenwaks S, Bar I (2008) Appl Phys Lett 93:041115
Shen YC, Lo T, Taday PF, Cole BE, Tribe WR, Kemp MC (2005) Appl Phys Lett 86:241116
Lo T, Gregory IS, Baker C, Taday PF, Tribe WR, Kemp MC (2006) Vibr Spectrosc 42:243–248
Shu J, Bar I, Rosenwaks S (2000) Appl Phys B 71:665–672
Daugey N, Shu J, Bar I, Rosenwaks S (1999) Appl Spectrosc 53:57–64
Wu DD, Singh JP, Yueh FY, Monts DL (1996) Appl Opt 35:3998–4003
De Lucia FC, Harmon RS, McNesby KL, Winkel RJ, Miziolek AW (2003) Appl Opt 42:6148–6152
Gronlund R, Lundqvist MSS (2006) Applied Spectroscopy 60853-859
Alden M, Westblom U, Goldsmith JEM (1989) Opt Lett 14:305–307
Huang YL, Gordon RJ (1992) J Chem Phys 97:6363–6368
Tserepi AD, Wurzberg E, Miller TA (1997) Chem Phys Lett 265:297–302
Summerlin LR, Borgford CL, Ealy JB (eds) (1988) Chemical demonstrations: a sourcebook for teachers, 2nd edn. American Chemical Society, Washington, DC
Luque J, Crosley DR (2000) J Phys Chem A 104:2567–2572
Kosmidis C, Marshall A, Clark A, Deas RM, Ledingham KWD, Singhal RP (1994) Rapid Commun Mass Spectrom 8:607–614
Arusi-Parpar T, Heflinger D, Lavi R (2001) Appl Opt 40:6677–6681
Boudreaux GM, Miller TS, Kunefke AJ, Singh JP, Yueh FY, Monts DL (1999) Appl Opt 38:1411–1417
Swayambunathan V, Sausa RC, Singh G (2000) Appl Spectrosc 54:651–658
Luque J, Crosley DR (1999) J Chem Phys 111:7405–7415
Lin MF, Lee YT, Ni CK, Xu SC, Lin MC (2007) J Chem Phys 126:064310
SenGupta S, Upadhyaya HP, Kumar A, Dhanya S, Naik PD, Bajaj P (2008) Chem Phys Lett 452:239–244
Sheaff CN, Eastwood D, Wai CM, Addleman RS (2008) Appl Spectrosc 62:739–746
Acknowledgments
This work was supported in part by the U.S. Army Research Office.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Arnold, B., Kelly, L., Oleske, J.B. et al. Standoff detection of nitrotoluenes using 213-nm amplified spontaneous emission from nitric oxide. Anal Bioanal Chem 395, 349–355 (2009). https://doi.org/10.1007/s00216-009-2990-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-2990-7