
Abstract
In this paper, the results obtained by five independent methods for the quantification of fumonisins B1, B2, and B3 in raw maize are reported. Five naturally contaminated maize samples and a reference material were analyzed in three different laboratories. Although each method was validated and common calibrants were used, a poor agreement about fumonisin contamination levels was obtained. In order to investigate the interactions among analyte and matrix leading to this lack of consistency, the occurrence of fumonisin derivatives was checked. Significant amounts of hidden fumonisins were detected for all the considered samples. Furthermore, the application of an in vitro digestion protocol to raw maize allowed for a higher recovery of native fumonisins, suggesting that the interaction occurring among analytes and matrix macromolecules is associative rather than covalent. Depending on the analytical method as well as the maize sample, only 37–68% of the total fumonisin concentrations were found to be extractable from the samples. These results are particularly impressive and significant in the case of the certified reference material, underlying the actual difficulties in ascertaining the trueness of a method for fumonisin determination, opening thus an important issue for risk assessment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
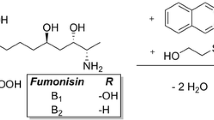
Fumonisins are a group of structurally related Fusarium mycotoxins produced mainly by Fusarium verticilloides and Fusarium proliferatum. Among them, fumonisins B1, B2, and B3 (FB1, FB2, and FB3, respectively) are the major mycotoxins produced in corn. These compounds are characterized by a 20 carbon aminopolyhydroxyalkyl chain diesterified with propane-1,2,3-tricarboxylic acid (tricarballylic acid; Fig. 1) [1, 2].

Main fumonisin structures

Comparison of the free FBs (sum of FB1, FB2, and FB3), total fumonisins found in the extract after hydrolysis (measured as HFBs and expressed as sum of FB1, FB2, and FB3 equivalents), and total fumonisins found in the samples after hydrolysis (measured as HFBs and expressed as sum of FB1, FB2, and FB3 equivalents) found in selected samples using method 1
Fumonisins may cause a variety of diseases in animals, as well as hepatocarcinogenic, hepatotoxic, nephrotoxic, and cytotoxic effects in mammals. Moreover, there is evidence of a high incidence of human esophageal cancer associated with FB1 exposure [3]. For these reasons, FB1 has been declared as a class 2B carcinogen by the International Agency for Research on Cancer. Recently, the European Union has enforced the new legislation for fumonisins in food: According to this regulation, the limits for total fumonisins in unprocessed maize (4,000 µg/kg), maize for direct human consumption (1,000 µg/kg), maize-based breakfast cereals and snacks (800 µg/kg), and in baby food (200 µg/kg) have been established (EC No. 1126/2007).
Analytical methods to detect the occurrence of fumonisins in corn-based products usually involve extraction with aqueous methanol or acetonitrile, purification on strong anion-exchange cartridges or immunoaffinity columns (IAC) and determination by RP-HPLC using fluorescence detection (FLD) and precolumn derivatization with ortho-phthalaldehyde (OPA) and 2-mercaptoethanol [4–6]. Although this method has been adopted by AOAC International as an official method for corn analysis [5], it has several disadvantages such as the instability of the derivatives and the fluorescent interferences caused by matrix components, making an accurate quantification very difficult, especially with processed corn foods [7–9]. For these reasons, in the last decade, a number of mass spectrometry-based methods for fumonisin detection have been proposed [10, 11]. As major advantage, no fumonisin derivatization is required, obtaining good recovery and sensitivity. Moreover, mass spectrometry-based methods allow to perform multiresidual determinations of a great number of mycotoxins in the same sample [12, 13].
The problem of fumonisin contamination is further complicated by the fact that hydrolyzed forms and degradation products such as N-carboxymethyl-fumonisins and N-(1-deoxy-d-fructos-1-yl) fumonisin B1 were found, although the latter was found in small amounts only in thermally treated products. More recently, other unidentified bound forms were detected in thermally treated food products such as cornflakes [14–16]. These forms are not directly detectable, but their presence has been inferred from the observation that, upon alkaline hydrolysis of the food matrix, the amount of released hydrolyzed fumonisins is often higher than that expected by the hydrolysis of the fumonisins detected in the sample using the routine procedure. The nature of this masking mechanism has been attributed to the formation of covalent bonds between the tricarballylic groups of fumonisins and the hydroxyl groups of starch or the amino or sulfidryl groups of the side chains of amino acids in proteins. The possibility of such an interaction has been proved as reacting fumonisins at high temperature with α-methyl-glucoside or protected amino acids as model systems for starch and proteins, respectively [14], although direct evidence of the presence of these derivatives has not been obtained until now in foods. The occurrence of hidden fumonisins was demonstrated by several authors [17–20] in corn-based foods by using HPLC-FLD and HPLC-MS. In particular, Dall’Asta et al. [19] showed that hidden fumonisins occurrence is not restricted to thermally treated products, but may be also found in mild-treated products such as corn flour and corn-based pasta. Thus, besides thermal effects [21], other masking mechanisms should be taken into account for the evaluation of the occurrence of hidden fumonisins in food.
Masking phenomena may occur already in the field between mycotoxins and plant macroconstituents, as a defensive response of the plant to the infection in order to allow the chemical and/or physical compartmentalization of the target molecule. As proved for deoxynivalenol-3-glucoside and zearalenone-4-glucoside, metabolites can be formed through conjugation [22, 23]. These conjugated metabolites are usually stable under extraction conditions, but when not properly monitored, can escape routine analysis.
Moreover, several compounds can be associated with protein or starch through non-covalent interactions [24]. Such complexations may be broken during the extraction process, on account of different experimental parameters applied during extraction such as solvent polarity, pH, ionic strength, time, and temperature, giving rise to the native form of the analyte. Thus, different extraction procedures will generally lead to different recoveries of the analytes.
Several authors reported poor recoveries for fumonisins from different food matrices, especially when naturally contaminated corn-based foods were analyzed [25–29]. Visconti et al. [4] reported a collaborative study involving 23 laboratories: Six samples were analyzed by using the same method based on IAC purification and HPLC analysis with fluorescence detection after OPA derivatization. The analyzed samples were one blank, two spiked corn samples, and three naturally contaminated samples. The collected data were consistent, proving the interlaboratory reproducibility and ruggedness of the chosen method. Nevertheless, no information about consistency of data obtained by different extraction procedures was given. Concerning the relation among hidden fumonisins and low free fumonisin recovery, Scott and Lawrence [29] underlined that, for corn-based food, distinction must be made between real losses, binding, and any matrix-related method problems. The fumonisin binding to matrix constituents could, thus, represent a problem for toxin quantification in real samples: If the nature of this binding is an associative phenomenon, the use of different analytical conditions may lead to different quantification results, although the methods used are validated.
The aim of this study is to compare the results obtained for naturally contaminated corn samples obtained by means of five independent analytical methods, which differ not only in the chosen analytical technique but also in the applied extraction procedure. Each method was previously validated using common calibrants, the same blank material, and the same reference material. The results were then compared in order to get more information about data consistency. Furthermore, hidden fumonisin occurrence was also evaluated for the sample set and related to free fumonisin results, trying to get an explanation for the low consistency found in interlaboratory comparison studies. In order to further investigate the matrix–analyte interactions, in vitro digestion experiments were performed on two selected raw maize samples and on a reference material [30]. The fumonisin levels obtained after matrix digestion were compared to those obtained for free fumonisins and after sample alkaline hydrolysis.
Materials and methods
Chemicals
Fumonisin B1, B2, and B3 standards (1 mg) were purchased from Biopure (Tulln, Austria). All solvents used (LC grade) were obtained from Carlo Erba (Milan, Italy); bidistilled water was produced in our laboratory utilizing an Alpha-Q system (Millipore, Marlborough, MA, USA). All salts were from Baker (Mallinckrodt Baker, Phillipsburg, NJ, USA), naphthalene 2,3-dicarboxyaldehyde (NDA) was from Sigma (Stuttgart, Germany). The reference material was a maize flour containing fumonisins B1 and B2, 2,406 ± 630 and 630 ± 116 µg/kg. MultiSep 211 Fum columns and AgraQuant Total Fumonisin Assay 0.25/5.0 Kit were from Romer Labs (Tulln, Austria). All chemicals for the digestive fluids were obtained from Sigma (Stuttgart, Germany).
Experimental design
In order to compare the results obtained by using independent analytical methods, six corn flours were chosen: one blank and five naturally contaminated samples. Each sample was mixed and split into aliquots (100 g) to be analyzed by the selected laboratories. The same standards were chosen to perform the calibration experiments. Calibration curves, repeatability, recovery, quantification, and detection limits of each method were obtained using the same blank matrix spiked with FB1, FB2, and FB3 at proper concentration levels.
Method 1 and the digestion assay were performed at the Food Chemistry Laboratory, Department of Organic Chemistry, University of Parma (Parma, Italy). Methods 2 and 3 were performed by different analysts and in different periods at the Christian Doppler Laboratory for Mycotoxin Research, Center for Analytical Chemistry, Department for Agrobiotechnology, BoKu University (Vienna, Austria). Methods 4 and 5 were performed by different analysts and in different periods at the Romer Labs Division Holding, Technopark 1, 3430 Tulln, Austria Romer Labs Division Holding (Tulln, Austria).
The analytical performances of the chosen methods are reported in Table 1.
Terminology
The terminology used in the present study to indicate fumonisin amounts found after different treatments is the following:
-
1.
Extractable fumonisins: the sum of FB1, FB2, and FB3 found in each sample upon application of common extraction conditions.
-
2.
Total fumonisins: the sum of FB1, FB2, and FB3 found in each sample after hydrolysis. This value is actually obtained by measuring HFB1, HFB2, and HFB3 and then converting the data into FBs equivalents by means of the proper conversion factor.
-
3.
Hidden fumonisins: the difference between total fumonisins and extractable fumonisins.
Hydrolyzed fumonisin preparation
Hydrolysis procedure was performed according to the method proposed by Dall’Asta et al. [20]. Briefly, a standard solution of the three fumonisins (50 µg/ml of each, 1 ml) was prepared in acetonitrile/water 1:1 (v/v) and evaporated to dryness. The residue was redissolved in 2 M KOH and allowed to react for 60 min. After hydrolysis, the mixture was extracted twice by liquid–liquid partition using ethyl acetate. The organic phases were pooled, evaporated under N2 stream, and redissolved in 1 ml acetonitrile/water (1:1). The conversion yield was quantitative, as already reported [20]. No partially hydrolyzed fumonisins were found. Calibration curves were prepared by proper dilution of the standard solution, assuming that the total conversion of the native compounds to be in the hydrolyzed forms.
Method 1
Sample preparation for the analysis of extractable fumonisins
Aliquots (5 g) of finely ground corn were blended in a high-speed blender (Ultraturrax T25, IKA, Stauffen, Germany) with 50 ml of water/methanol (30:70, v/v) for 10 min at 6,000 rpm and then stirred for 50 min. After centrifugation at 3,500 rpm for 15 min, the extract (2 ml) was filtered through a 0.45-µm nylon filter and analyzed by LC–MS/MS as described below.
Sample preparation for the analysis of total fumonisins
Aliquots (5 g) of finely ground corn were blended in a high-speed blender (Ultraturrax T25, IKA, Stauffen, Germany) with 50 ml of 2 M KOH for 10 min at 6,000 rpm and stirred for 50 min. Then, 50 ml of acetonitrile was added, and after stirring for 10 min, the two layers were separated by centrifugation at 3,500 rpm for 15 min. The extract (2 ml) was evaporated to dryness under a stream of nitrogen, and the residue was redissolved in water/methanol 30:70 (v/v), filtered through a 0.45-µm nylon filter, and analyzed by LC–MS/MS as described below. Fumonisins obtained after sample hydrolysis were measured as the sum of HFB1, HFB2, and HFB3. All the results are expressed as the sum of FB1, FB2, and FB3 equivalents, considering a correction factor due to the different molecular weight of native and hydrolyzed compounds and referred to as “total fumonisins”. Hidden fumonisins were calculated as the difference between total fumonisins and free fumonisins.
LC–MS/MS analysis
The LC–electrospray ionization (ESI)–MS/MS analysis was performed as reported by Dall’Asta et al. [19, 20]. Briefly, a 2695 Alliance separation system (Waters Co., Milford, MA, USA) equipped with a QuattroTM triple quadrupole mass spectrometer with an electrospray source (Micromass, Waters, Manchester, UK) was used. Chromatographic conditions were the following: column, C18 XTerra Waters narrow bore (250 mm × 2.1 mm, 5 µm) equipped with a C18 precolumn cartridge; flow rate, 0.2 ml/min; column temperature, 30 °C; injection volume, 10 µl. Gradient elution was performed using water (eluent A) and methanol (eluent B), both acidified with 0.1% formic acid: 0–2 min, isocratic step at 70% A; 2–5 min to 45% B; 5–25 min to 90% B; 25–35 min isocratic step at 90% B; finally, a reequilibration step at 70% A (initial conditions) for 15 min was performed (total analysis time, 50 min). MS parameters: ESI+ (positive ion mode); capillary voltage, 4 kV; cone voltage, 50 V for FBs and 30 V for HFBs; extractor voltage, 2 V; source block temperature, 120 °C; desolvation temperature, 350 °C; desolvation and cone gas (nitrogen) 600 and 50 l/h, respectively. Detection was performed using a multiple reaction monitoring (MRM) mode by monitoring two transitions for each analyte (collision energy, 35 eV for FBs and 20 eV for HFBs). In particular, the monitored transitions were as follows: FB1 722.4 → 334.4, 722.4 → 352.4; FB2 and FB3 706.4 → 318.4, 706.4 → 336.4; HFB1 406.5 → 370.5, 406.5 → 388.5; HFB2 and HFB3 390.5 → 336.4, 390.5 → 372.5. The first transition reported was used for quantification, while the second transition was chosen as qualifier.
Method 2
Sample preparation for the analysis of extractable fumonisins
The extraction of fumonisins was performed as above. The samples were then analyzed following the mass spectrometric method reported below.
Sample preparation for the analysis of total fumonisins
The extraction of total fumonisins from raw corn was performed as already described. Moreover, in order to check whether hidden forms occurred in the extract, an aliquot of the extract prepared for free FBs analysis was hydrolyzed. Briefly, an aliquot of extract (20 ml) obtained as above was evaporated, redissolved in 50 ml 2 M KOH, and stirred for 50 min. After addition of acetonitrile (50 ml), the solution was stirred for 10 min; then, the organic layer was separated by centrifugation at 3,500 rpm for 15 min. The extract (2 ml) was then evaporated to dryness under a stream of nitrogen, and the residue redissolved in water/methanol 30:70 (v/v), filtered through a 0.45-µm nylon filter, and analyzed by LC–MS/MS.
LC–MS/MS analysis
An 1100 Series HPLC System (Agilent, Waldbronn, Germany) in conjunction with a QTrap LC–MS/MS System (Applied Biosystems, Foster City, USA) equipped with an ESI source was used for analysis. Chromatographic conditions were the following: column, C18 Hypersil (Thermo, 150 mm × 2.1 mm, 5 µm); flow rate, 0.6 ml/min; column temperature, 25 °C; injection volume, 10 µl. Gradient elution was performed using water (eluent A) and acetonitrile (eluent B), both acidified with 0.2% formic acid: 0–1 min, isocratic step at 80% A; 1–6 min to 100% B; 6–9 min isocratic step at 100% B; finally, a reequilibration step at 80% A (initial conditions) for 4 min was performed (total analysis time, 13 min). MS parameters: Turbo Spray positive; declustering potential (DP) and collisional energy (CE) were optimized for each analyte; source block temperature, 425 °C. Detection was performed using a MRM mode by monitoring three transitions for each analyte: The main transition was used for quantification, while the other transitions were chosen as qualifiers. In particular, the monitored transitions were as follows: FB1 722.2 → 334.3 (CE 51 V, DP 76 V), 722.2 → 704.3 (CE 31 V, DP 76 V), 722.2 → 352.3 (CE 39 V, DP 76 V); FB2 and FB3 706.2 → 336.5 (CE 40 V, DP 68 V), 706.2 → 318.4 (CE 47 V, DP 68 V), 706.2 → 354.4 (CE 37 V, DP 68 V); HFB1 406.2 → 388.3 (CE 19 V, DP 36 V), 406.2 → 370.3 (CE 21 V, DP 36 V), 406.2→352.3 (CE 21 V, DP 36 V); HFB2 and HFB3 390.2 → 336.4 (CE 24 V, DP 41 V), 390.2 → 372.4 (CE 20 V, DP 41 V), 390.2 → 354.4 (CE 21 V, DP 41 V).
Method 3
Sample preparation
Sample (0.5 g) was added with 2 ml of extraction solvent (acetonitrile/water/acetic acid 79:20:1, v/v/v). The samples were extracted for 90 min using a GFL 3017 rotary shaker (GFL, Burgwedel, Germany) and subsequently centrifuged for 2 min at 3,000 rpm (radius 15 cm) on a GS-6 centrifuge (Beckman Coulter Inc., Fullerton, CA, USA). The extracts were transferred into glass vials using Pasteur pipettes, and 350-μl aliquots were diluted with the same volume of dilution solvent (acetonitrile/water/acetic acid 79:20:1, v/v/v). After appropriate mixing, 5 μl of the diluted extract was injected into the LC–MS/MS system without further pretreatment.
LC–MS/MS analysis
Fumonisin detection was achieved using the multitoxin method described by Sulyok et al. [12]. Briefly, detection and quantification were performed with a QTrap 4000 LC–MS/MS System (Applied Biosystems, Foster City, CA, USA) equipped with a TurboIonSpray ESI source and an 1100 Series HPLC System (Agilent, Waldbronn, Germany). Chromatographic separation was performed at 25 °C on a Gemini® C18 column, 150 × 4.6-mm i.d., 5-μm particle size, equipped with a C18 4 × 3-mm i.d. security guard cartridge (all from Phenomenex, Torrance, CA, USA). Both eluents contained 5 mM ammonium acetate and were composed of methanol/water/acetic acid 10:89:1 (v/v/v; eluent A) or 97:2:1 (eluent B), respectively. After an initial time of 2 min at 100% A, the proportion of B was increased linearly to 100% within 12 min, followed by a hold time of 3 min at 100% B and 4-min column reequilibration at 100% A. The flow rate was 1 ml min−1. ESI–MS/MS was performed in the MRM mode both in positive and negative polarities in two separate chromatographic runs per sample by scanning two fragmentation reactions per analyte with the following settings: source temperature 550 °C, curtain gas 10 psi (69 kPa of max. 99.5% nitrogen), ion source gas 1 (sheath gas) 50 psi (345 kPa of nitrogen), ion source gas 2 (drying gas) 50 psi (345 kPa of nitrogen), ion spray voltage −4,000 and +4,000 V respectively, collision gas (nitrogen) high. Concerning fumonisins, declustering potential was set as follows: 91 V for FB1, 96 V for FB2 and FB3. The main transitions used for quantification were 722.5 → 334.4 (CE 57 V) for FB1 and 706.3 → 336.3 (CE 59 V) for FB2 and FB3; the qualifier transitions were 722.5 → 352.3 (CE 55 V) for FB1 and 706.3 → 318.5 (CE 51 V) for FB2 and FB3.
Method 4
Sample preparation
For fumonisin analysis, 25 g of ground sample was extracted with 100 ml water–acetonitrile (1:1, v/v) by shaking for 1 h. After filtration, the supernatant was adjusted to pH 6–9 with 0.5 N NaOH for allowing a good recovery during the purification step. Afterward, 3 ml of extract was placed into a 16 × 125-mm test tube, and 8 ml of methanol–water (3:1, v/v) was added to the sample. The purification step was performed using a MultiSep 211 Fum column, preconditioned with 5 ml of methanol followed by 5 ml of methanol–water (3:1, v/v) solution.
The diluted sample was passed through the column, which was then rinsed with 8 ml of methanol–water (3:1, v/v) followed by 3 ml of methanol. Fumonisins were eluted with 10 ml of methanol–acetic acid (99:1, v/v). The eluate was then evaporated to dryness under vacuum in a 60 °C water bath. After reconstitution with 1 ml of methanol, the sample was added with 1 ml sodium borate buffer (0.05 M, pH 9.5), 0.5 ml sodium cyanide reagent (13 mg sodium cyanide in 100 ml water) and 0.5 ml NDA reagent (25 mg NDA in 100 ml methanol). After heating for 20 min in a 60 °C water bath and cooling at 8 °C for 4 min, the sample was diluted with 7 ml of phosphate buffer (0.05 M, pH 7.4)/acetonitrile (40:60, v/v) and analyzed by HPLC with fluorescence detection.
HPLC-FLD analysis
The analysis was performed using a Shimadzu SCL 10A system controller, equipped with a RF-10Axl fluorescence detector, a SIL-10A autoinjector, and a LC-10AD pump. Chromatographic conditions were the following: column, Brownlee C18 (100 mm × 4.6 mm, 5 µm); flow rate, 0.2 ml/min; injection volume, 80 µl. An isocratic elution was performed using water–acetonitrile–acetic acid 52:47:1, v/v/v. Fluorescence detection was performed at λ ex = 420 nm and λ em = 500 nm.
Method 5
Sample preparation and analysis
For fumonisin analysis, 10 g of ground sample was extracted with 50 ml methanol–water (70:30, v/v) by blending for 3 min. After filtration, the supernatant was adjusted to pH 7–8 with 0.5 N NaOH, if necessary. Afterward, 50 µl of extract was diluted with 950 µl of water and analyzed by ELISA kit, according to the AgraQuant Total Fumonisin Assay Protocol (Method PI-000082-1).
Digestion experiments
The digestion experiment was performed according to the protocol reported by Versantvoort et al. [31]. Briefly, the main digestive juices were prepared by mixing the proper amount of salts and enzymes, according to the original procedure, and heated to 37 °C before use. The digestion started by adding 3 ml saliva to 2 g contaminated sample and was incubated for 5 min. Then, 6 ml of gastric juice was added, and the mixture was stirred for 2 h. Finally, 6 ml of duodenal juice, 3 ml bile, and 1 ml bicarbonate solution (1 M) were added simultaneously, and the mixture was stirred again for 2 h. At the end of the in vitro digestion process, the digestion tubes were centrifuged for 5 min at 2,750×g, yielding the chyme (the supernatant) in which the concentration of mycotoxin was determined and the digested matrix (the pellet). The chyme was then purified using Sep-Pak C18 columns (Waters Co., Milford, MA, USA): The sample was applied to the cartridge, which was precontioned with 2 ml methanol followed by 2 ml bidistilled water. After a washing step (3 ml bidistilled water), fumonisins were eluted using 2 ml acetonitrile/water solution (1:1). The eluate was then analyzed by LC/MS/MS for both native and hydrolyzed forms. The occurrence of partially hydrolyzed fumonisins was also checked.
Results and discussion
Comparison of the chosen methods
In order to compare the results obtained for naturally contaminated corn samples, five independent analytical methods for fumonisins were chosen. In particular, selected methods differed not only in the analytical technique but also in the applied extraction procedure: All the tested protocols were previously published and used for routine analysis in the involved laboratories after in-house validation. Moreover, the most common analytical techniques for mycotoxin detection were covered: Methods 1 and 2 were based on the same procedure but performed in different laboratories using different chromatographic conditions and different instruments; method 3 is the well-known multimycotoxin method based on mass spectrometry reported by Sulyok et al. [12]; method 4 involved fluorescence detection after a derivatization step performed using a commercial kit; finally, method 5 was an ELISA test also performed using a commercial kit. Selected raw maize samples (n = 5) were prepared for interlaboratory analysis by grinding and mixing before sampling; the sub-samples were stored at +4 °C until fumonisin determination.
Before the experiment, each method was in-house-validated using common calibrants, and the method performances were evaluated, as reported in Table 1. The same blank material was checked and used for matrix-matched calibration. Moreover, a reference material (raw maize) was analyzed, and the results were compared in order to get more information about data consistency. Results showed a good agreement.
The five selected raw maize samples were analyzed in replicate and the results were compared, as reported in Table 2. Since the number of replicates was different between laboratories, we decided not to include the standard deviation of measurements. However, CV of measurements were in agreement with method precision as obtained for in-house validations, which was also reported in Table 2. The occurrence of HFBs in raw maize was also checked by methods 1 and 2: None of the selected samples was found to be positive for these analytes.
Surprisingly, the obtained data showed a very low consistency: The FB values found for each sample were very different. Also, the data obtained using methods 1 and 2 were different, although obtained using very similar conditions.
Since different methods were used in different laboratories, we actually cannot differentiate between method and laboratory bias. However, each method was thoroughly in-house-validated, and the same calibrants were used in all laboratories. Moreover, the sample stability and homogeneity was checked by including a reference material in the considered sample set. Thus, we assume that laboratory bias is not the reason for the different measured fumonisin levels.
These unexpected results pointed out a serious analytical problem in fumonisin determination, as already reported by several authors. These recovery problems cannot be due to poor sample preparation since the procedures used in this study were fully validated, performed by trained operators, and commonly accepted. A possible explanation can be found in the method’s ability to extract fumonisins from the matrix, but in this case, the same method should always over- or underestimate the analyte on account of the conditions applied to the extraction and/or purification step (solvent, pH, temperature, and time): In our data, no systematic trend can be found.
Another possible reason may be actually ascribed to the occurrence of hidden fumonisins, which have been already reported for thermal-treated corn-based products by several authors [14, 17–20]. These masked derivatives can be co-extracted together with the free forms, causing a misidentification of the real contamination level.
Hidden fumonisin determination
The occurrence of hidden fumonisins in raw maize was checked by alkaline hydrolysis, as reported by Dall’Asta et al. [20]. In particular, the sample was hydrolyzed according to the procedure reported in the “Materials and methods” section for total FBs quantification. Hidden fumonisins were calculated as the difference between extractable FBs and total FBs values. Moreover, the extract was treated under alkaline conditions in order to check whether hidden forms were co-extracted together with commonly detectable forms. The collected data are reported in Table 3. Extractable fumonisins, total fumonisins found in each extract after hydrolysis and total fumonisins found in each sample after hydrolysis were compared in Fig. 2. All the data were obtained using Method 1.
All the selected samples were found to contain hidden fumonisins in concentration comparable to extractable fumonisins. To our knowledge, this is the first report about the occurrence of hidden fumonisins in raw maize, although these forms were already detected in corn flour [19].
Since these raw maize samples did not undergo any thermal treatment, this result is of great concern, suggesting that the fumonisin masking mechanism is thus exerted also by the plant, as already described for other Fusarium mycotoxins such as deoxynivalenol and zearalenone [22, 23].
Since hidden fumonisins are released under alkaline hydrolysis, the masking mechanism probably involves the tricarballylic side chains of the molecule, as suggested by several authors [14, 18]. However, the in vitro model proposed by Seefelder [14] to explain the fumonisin covalent binding to protein or starch involved an intramolecular activation of the carboxylic moieties through a cyclic anhydride formation, which can be obtained only by heating. This mechanism is thus possible only for thermal-treated products, but cannot explain the binding occurring in raw maize, which is rather probably due to a physical entrapment eventually obtained via specific or non-specific enzymatic activity. As far as the maize macromolecular components are involved in these interactions, in several studies, hidden fumonisins have been mainly found associated to protein fraction [17, 18]. Proteins in corn are mainly represented by zeins, which are water-insoluble storage proteins showing a helical character, made up of nine helical segments of about 20 amino acids with glutamine-rich turns or loops [24, 32, 33]. Inside this tridimensional structure, zein may host several guests such as xanthophylls [24]. On the other hand, the decrease in fumonisin concentration in cornstarch and cornmeal after spiking was also described [27], suggesting a possible interaction of the target toxin with starch. In particular, only 43% of C-14-labeled FB1 materials extracted from cornstarch were recovered by immunoaffinity purification, and 50% remained after 30 days in corn meal. Since no thermal treatment was applied to these matrices, the binding of labeled fumonisin to proteins or starch may be due to enzymatic activity or to other unknown mechanism.
A possible explanation of these phenomena may be the toxin complexation into supramolecular structures formed by macromolecules such as starch and zeins: this hypothesis is strongly supported by the presence of hidden fumonisins in the extract obtained with the extraction solvents used for fumonisin determination. This observation is potentially explained by the presence of maize prolamin proteins in the extract: Indeed, the solvent used to extract fumonisins from the food matrix is also able to dissolve the prolamin (zein) fraction of maize. These protein bodies retain their tertiary structure in solution, thus potentially masking the presence of hidden fumonisins, which cannot be directly determined. Enzymatic digestion lead to the declustering of these structures, thus releasing the entrapped FBs. Alkaline hydrolysis is also able to destabilize these supramolecular interactions as probably the HFBs did not yet possess the structural features that allow the formation of such strong complexes.
This association can be considered as a physical entrapment, which does not allow the complete recovery of the target compound during analysis. When such associations occur in the matrix, the ratio among extractable and retained fumonisins is influenced by physicochemical parameters such as pH, solvent polarity, temperature, and time. Thus, different extraction methods can lead to different recovery from the same sample, even when fully validated methods are used. During validation, recovery is actually evaluated by spiking experiments at different levels: Although this procedure is suitable to evaluate possible critical points in the procedure such as analyte losses during extraction or clean up, the interaction among matrix constituents and analyte occurring in naturally contaminated samples leading to analyte masking effects cannot be compensated.
Simulated digestion of raw maize
In order to unravel this intricate problem, we applied a different approach, based on the use of a digestion protocol proposed by Versantwoort and Omen for food contaminant bioavailability assessment [31]: Indeed, digestion of the matrix would reveal if fumonisins are bound to proteins or starch through covalent binding or by complexation as only in the latter case free native fumonisins should be released.
At first, the analyte stability under applied conditions was confirmed by applying the digestion protocol to a fumonisin standard solution. Then, fumonisin-contaminated raw maize was treated as described by the protocol, and FBs were quantified in chyme after digestion and compared to the FBs concentration found in non-digested sample. The protocol was applied to two highly contaminated raw maize samples and to the same reference material used in this study for method comparison. The results were compared to those obtained for extractable fumonisins (method 1) and total fumonisins after alkaline hydrolysis (method 1), as reported in Fig. 3.

Level comparison of free FBs (expressed as the sum of FB1, FB2, and FB3; method 1), total FBs after hydrolysis (measured as HFBs and expressed as the sum of FB1, FB2, and FB3 equivalents; method 1), and free FBs (sum of FB1, FB2, and FB3) after digestion found in raw maize samples (F, G) and in reference material (RM)
An increase of 30–50% in fumonisins concentration after digestion was measured, suggesting that the binding mechanism of fumonisins in raw maize is mainly due to an association with macromolecules such as starch and proteins. In addition, no hydrolyzed or partially hydrolyzed fumonisins were found in the chyme. Finally, the total fumonisin levels obtained after digestion are slightly higher than those obtained after alkaline hydrolysis, suggesting that hidden fumonisins are actually native forms, which can be released under digestive conditions.
These results are particularly impressive in the case of the reference material as, in front of a declared fumonisin contamination of 3,036 ± 746 µg/kg, the real amount of free fumonisins detected upon digestion is as high as 8,010 ± 426 µg/kg. These data open an interesting issue for method validation: Hidden fumonisins may occur also in reference materials since this sample is usually naturally contaminated, but the reference values do not usually consider this issue. An “in-house” validated method should confirm the reference value, showing thus a good reliability when compared to reference data. On the other hand, the occurrence of higher level of fumonisins in reference material after digestion showed that the reference data are not the true data: Measuring accuracy in in-house validation does not actually mean measuring trueness. This is a major point to be considered during method validation in order to avoid significant underestimation of fumonisin contamination in maize.
A very similar approach was already used for evaluating in vitro digestion effects on hidden fumonisins in cornflakes [30], without finding a significative contribution of hidden fumonisins in chyme. Since cornflakes are thermally treated products, the covalent binding of fumonisins to matrix macroconstituents could occur, and in this case, native forms could not be released upon digestion. Further experiments should be performed in order to investigate the occurrence of associative or covalent binding in processed corn-based products.
These results, although very preliminary and to be confirmed with a larger number of different samples, are of great concern for food safety and may offer an explanation to the so-called fumonisin paradox [34]: When such association occur between fumonisin and matrix macroconstituents, the common analytical methods, although well validated, are unable to detect the true level of mycotoxin in corn, giving thus an underestimation of the exposure to the target mycotoxin.
The terminology issue
So far, the terms “hidden fumonisins” [17, 18] and “bound fumonisins” [14] were indifferently used in the literature to indicate those fumonisin forms, which were not extractable by common procedures but which may release the native or the hydrolyzed forms upon digestion, thus exerting a toxic effect.
Moreover, those compounds obtained from fumonisins through a Maillard-type reaction involving reducing sugars were often referred as bound fumonisins, although these derivatives cannot release the native forms under gastric conditions.
This paper showed that hidden fumonisins may occur also in raw maize, and the reported data suggested an associative mechanism rather than a covalent binding as a possible explanation of this phenomenon. Thus, a clarification is needed on the terminology used for non-covalently bound and for covalently bound fumonisins.
Fumonisins can occur in maize as free forms (native, partially hydrolyzed, or totally hydrolyzed), as covalently bound fumonisins (i.e., to starch, proteins, and reducing sugars), and as non-covalently bound fumonisins. In our opinion, the term bound fumonisins should be used only for those compounds, which involve a covalent linkage among the analyte and any matrix constituents. This linkage may involve the fumonisin free amino group or the carboxylic moieties. The amino derivatives cannot be hydrolyzed upon digestive conditions and cannot be released after alkaline hydrolysis; thus, they should be directly measured in the sample. Moreover, their toxicity should be carefully evaluated because the covalent binding could actually act as a detoxification way. On the other hand, hydrolyzed fumonisins could be released from the carboxy derivatives in the gastrointestinal tract: These forms should be indirectly measured by sample alkaline hydrolysis.
On the other hand, the term hidden fumonisins should be used only for non-covalently bound derivatives, which are formed through an associative interaction between fumonisins and matrix macroconstituents. Since this association is strongly dependent upon the environmental conditions, digestive processes may release the native forms, still exerting their own toxicity. This masking mechanism should be further studied, in order to investigate if the association is driven by specific or aspecific phenomena exerted by plant upon mold contamination. In any case, hidden fumonisins may occur in raw maize as well as in processed products.
Finally, as extractable fumonisins, we can define the non-covalently bound FBs, which might actually be partially released by common extraction procedures on account of the applied extraction conditions. Thus, this amount obviously depends on the applied method, as proved by this report. The common methods, although thoroughly validated, cannot give an absolute measurement of the fumonisin contamination since the analysis outcome is dominated by the extraction conditions. Strong efforts are thus urgently needed to develop an analytical procedure for measuring with a high trueness degree the real fumonisin content in maize.
Conclusion
Fumonisin quantification in maize and corn-based products is generally regarded as difficult as low comparabilities of results were often described in the literature. Some interlaboratory studies for method validation have been performed so far, but none of them evaluated common samples by means of different analytical procedures. In this study, several raw maize samples were measured using different extraction protocols and different analytical detections. Although each method was in-house-validated and common calibrators were used, a poor agreement of fumonisin contamination levels was obtained. In order to investigate possible reasons for this lack of consistency, the binding of the toxins to matrix components by covalent or non-covalent interactions was checked. Bound fumonisins released either under alkaline conditions or after digestion from all the considered samples were at levels comparable or even higher than those measured without extra sample treatment. This is the first report about the occurrence of bound fumonisins in raw maize, suggesting that the masking effect is at least partly exerted by the mold infected plant, as already reported for other mycotoxins, rather than being induced by thermal treatment during maize processing. Furthermore, the application of an in vitro digestion protocol to raw maize allowed for a higher recovery of native fumonisins, suggesting that the interaction occurring among analytes and matrix macromolecules is associative rather than covalent.
The difficulties encountered in fumonisin recoveries by the application of common analytical methods are thus additionally hampered by hidden fumonisins due to different extraction efficiencies. Recovery evaluation during in-house validation cannot actually be performed by spiking blank matrices since the added analytes do not undergo the same interactions with matrix components as fumonisins produced after fungal infection in the field. Even with the use of naturally contaminated maize as reference material, the recovery evaluation is critical as different maize samples can contain different amounts of hidden fumonisins. Thus, a method which is suited to quantify the “true” content of free and bound fumonisins through an effective matrix disaggregation is strongly required in order to avoid significant underestimation of mycotoxin levels.
Moreover, the masking effect exerted on fumonisins by maize macromolecules such as starch and protein through associative interactions should be further investigated since the occurrence of masked native fumonisins in raw material may be a probable explanation of the so-called fumonisin paradox.
References
Murphy PA, Hendrich S, Hopmans EC, Hauck CC, Lu Z, Buseman G, Munkvold G (1996) Adv Exp Med Biol 392:323–334
Sewram V, Mshicileli N, Shephard G, Vismer HF, Rheeder JP, Lee Y-W, Leslie JF, Marasas WFO (2005) J Agric Food Chem 53:4861–4866
Creppy EE (2002) Toxicol Lett 127:19–28
Visconti A, Boenke A, Solfrizzo M, Pascale M, Doko MB (1996) Food Addit Contam 13:929–938
Sydenham EW, Shephard GS, Thiel PG (1992) J AOAC Int 75:313–318
Solfrizzo M, De Girolamo A, Visconti A (2001) Food Add Contam 18:227–235
Visconti A, Solfrizzo M, De Girolamo A (2001) J AOAC Int 84:1828–1836
Avantaggiato G, De La Campa R, Miller JD, Visconti A (2003) J Food Prot 66:1873–1878
Scott PM, Lawrence GA (1996) Food Add Contam 13:823–832
Zoellner P, Mayer-Helm B (2006) J Chromatogr A 1136:123–169
Sforza S, Dall'Asta C, Marchelli R (2006) Mass Spectrom Rev 25:54–76
Sulyok M, Berthiller F, Krska R, Schuhmacher R (2006) Rapid Commun Mass Spectrom 20:2649–2659
Sulyok M, Krska R, Schuhmacher R (2007) Food Add Contam 24:1184–1195
Seefelder W, Knecht A, Humpf H-U (2003) J Agric Food Chem 51:5567–5573
Shier WT, Abbas HK, Abou-Karam M, Badria FA, Resch PA (2003) Fumonisins: abiogenic conversions of an environmental tumor promoter and common food contaminant. J Toxicol-Toxin Rev 22:591–616
Shier WT, Abbas HK, Badria FA (1997) J Nat Toxins 6:225–242
Kim E-K, Scott PM, Lau BP-Y (2003) Food Add Contam 20:161–169
Park JW, Scott PM, Lau BP-Y, Lewis DA (2004) Food Add Contam 21:1168–1178
Dall'Asta C, Galaverna G, Aureli G, Dossena A, Marchelli R (2008) World Mycotoxin J 1:1–10
Dall'Asta C, Galaverna G, Mangia M, Sforza S, Dossena A, Marchelli R (2009) Mol Nutr Food Res 53:492–499
Humpf HU, Voss KA (2004) Mol Nutr Food Res 48:255–269
Berthiller F, Dall'Asta C, Schuhmacher R, Lemmens M, Adam G, Krska R (2005) J Agric Food Chem 53:3421–3425
Schneweis I, Meyer K, Engelhardt G, Bauer J (2002) J Agric Food Chem 50:1736–1738
Momany FA, Sessa DJ, Lawton JW, Selling GW, Hamaker SAH, Willett JL (2006) J Agric Food Chem 54:543–547
De Girolamo A, Solfrizzo M, Visconti A (2001) J Food Prot 64:701–705
De Girolamo A, Solfrizzo M, von Holst C, Visconti A (2001) Food Add Contam 18:59–67
Kim EK, Scott PM, Lau BPY, Lewis DA (2002) J Agric Food Chem 50:3614–3620
Lawrence JF, Niedzwiadek B, Scott PM (2000) J AOAC Int 83:604–611
Scott PM, Lawrence GA (1994) J AOAC Int 77:541–545
Motta EL, Scott PM (2007) Mycotoxin Res 23:166–172
Versantvoort CHM, Oomen AG, Van de Kamp E, Rompelberg CJM, Sips A (2005) Food Chem Toxicol 43:31–40
Matsushima N, Danno G, Takezawa H, Izumi Y (1997) Biochim Biophys Acta 1339:14–22
Danzer LA, Ades H, Rees ED (1975) Biochim Biophys Acta 386:26–31
Shier WT (2000) J Toxicol-Toxin Rev 19:161–187
Acknowledgments
The authors thank the Christian Doppler Research Society and the Lower Austrian Government for their financial support. The authors also thank the Network Laboratory SITEIA (Sicurezza, Tecnologie, Innovazione Agroalimentare), Emilia Romagna Region, for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dall’Asta, C., Mangia, M., Berthiller, F. et al. Difficulties in fumonisin determination: the issue of hidden fumonisins. Anal Bioanal Chem 395, 1335–1345 (2009). https://doi.org/10.1007/s00216-009-2933-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-2933-3