
Abstract
Finite element method calculations were carried out to determine extinction spectra and the electromagnetic (EM) contributions to surface-enhanced Raman spectroscopy (SERS) for 90-nm Au nanoparticle dimers modeled after experimental nanotags. The calculations revealed that the EM properties depend significantly on the junction region, specifically the distance between the nanoparticles for spacings of less than 1 nm. For extinction spectra, spacings below 1 nm lead to maxima that are strongly red-shifted from the 600-nm plasmon maximum associated with an isolated nanoparticle. This result agrees qualitatively well with experimental transmission electron microscopy images and localized surface plasmon resonance spectra that are also presented. The calculations further revealed that spacings below 0.5 nm, and especially a slight fusing of the nanoparticles to give tiny crevices, leads to EM enhancements of 1010 or greater. Assuming a uniform coating of SERS molecules around both nanoparticles, we determined that regardless of the separation, the highest EM fields always dominate the SERS signal. In addition, we determined that for small separations less than 3% of the molecules always contribute to greater than 90% of the signal.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Surface-enhanced Raman spectroscopy (SERS) [1–3] is one of the most sensitive methods for obtaining vibrational spectra of molecules. The mechanism of SERS is primarily electromagnetic (EM) [4–7], where Raman scattering is enhanced by 105–106 by adsorbing molecules on a rough metal surface, typically Ag, but also Au and Cu. In some cases, even greater enhancements are possible which allows for the detection of individual molecules using single-molecule SERS (SMSERS) [8, 9] (often in these cases resonant Raman scattering and chemical effects contribute to the large enhancements [6, 10]). The large EM enhancements arise from localized surface plasmon resonances (LSPRs) excited on the surface of the metal. This excitation is from the incident light at frequency ω, \( \left| {\vec{E}\left( \omega \right)} \right|^2 \), as well as light emitted by the oscillating dipole induced in the molecule at the Stokes shifted frequency \( \omega \prime \), \( \left| {\vec{E}\left( {\omega \prime } \right)} \right|^2 \). Kerker et al. [11] demonstrated that the overall EM enhancement is proportional to \( \left| {\vec{E}\left( \omega \right)} \right|^2 \left| {\vec{E}\left( {\omega \prime } \right)} \right|^2 \), which is approximately \( \left| {\vec{E}\left( \omega \right)} \right|^4 \) (hereon denoted as \( \left| {\vec{E}} \right|^4 \)) if the width of the LSPR resonance is large compared to the difference in ω and \( \omega \prime \) (as is often the case).
SERS finds many applications, one being the detection of biomolecules using Ag or Au nanoparticles as SERS nanotags [12]. The nanotags are made by coating the nanoparticles with label molecules which have a known SERS spectrum, encapsulating them in a protective shell (e.g., SiO2), and functionalizing the surface to bind to a target analyte. SERS nanotags are interesting from a theoretical standpoint because their structure is simple, making it possible to address important fundamental questions about SERS, such as what structures generate the highest maximum and average EM enhancements. The first question is important for SMSERS, whereas the latter is important for the detection of low concentrations of molecules. To help answer such questions, computational electrodynamics methods, such as the discrete dipole approximation (DDA) [13, 14], the finite-difference time-domain (FDTD) method [15, 16], and the finite element method (FEM) [17] are often employed. Most attention has been focused on maximizing EM enhancements, where values greater than 108 have been calculated for single nanoparticles [18] and 1010 for coupled nanoparticles [19–21]. However, there have only been a few theoretical studies addressing another important question of how many molecules contribute to the SERS signal for a given structure [22].
Herein, we use FEM to study the EM contributions to SERS, focusing on nanotags composed of 90-nm-diameter Au nanoparticle dimers with a 20-nm SiO2 protective shell. This work expands on previous theoretical studies of strongly coupled nanowires [23] and spheres [21, 23–26]. We first study the extinction spectra, where we find that separations of less than 1 nm produce plasmon resonances that are strongly red-shifted from the 600 nm LSPR maximum associated with a Au monomer. Experimental transmission electron microscopy (TEM) images and correlated LSPR images—LSPR spectra are presented that confirm this behavior. We then use FEM to study EM enhancements for a variety of nanoparticle separations, including the possibility of touching or coalesced (partially fused) nanoparticles. From these results, we determine the conditions for SMSERS, and when many molecules are present what fraction contributes to the SERS signal for various molecular diameters and nanoparticle separations.
An important feature of the present study is that by using FEM we are able to calculate EM enhancements with much greater accuracy than is obtainable using DDA or FDTD. The latter methods suffer from stair-casing errors, and also make serious approximations to the EM fields at metal/dielectric interfaces. As a result, the EM enhancements obtained in past studies have often been subject to several order-of-magnitude uncertainties. Of course, even the FEM calculations are subject to errors associated with the use of a local dielectric constant; however, within this limitation, the present calculations provide fully converged results.
Materials and methods
Experimental
SERS nanotags composed of aggregated 90-nm-diameter gold spheres coated with 50-nm SiO2 were used as received from Oxonica Materials, Inc. TEM measurements were performed on Cu TEM grids coated with a 50-nm thick film of formvar and a 2–3 nm layer of amorphous C (Ted Pella). The nanotags were deposited on the TEM grid by drop-casting a 10-µL aqueous solution. TEM images were obtained on a JEOL JEM-2100F Fast TEM operating at 200 kV. LSPR spectra of individual nanotags were measured on an inverted microscope (Nikon TE300) using white-light illumination through a dry dark-field condenser (Nikon, numerical aperture (NA) = 0.7–0.95). Scattering from the sample was collected through an oil-immersion objective equipped with a variable NA iris set to NA = 0.5 (Nikon, Plan Fluor, 100X, oil, iris) onto a 1/3-m monochromator containing a low-dispersion grating blazed at 500 nm (150 groove/mm), and detected by a LN2-cooled CCD camera (Princeton Instruments Spec-10 400BR). Individual diffraction-limited spots were centered on the entrance slit of the spectrograph and LSPR spectra were collected from λ = 400 to 900 nm with a typical acquisition time of 3 s.
Finite element method (FEM)
FEM has been described in detail elsewhere [17]. Calculations were performed using an open-source FEM code, JFEM2D [27], to solve the frequency-domain scalar wave equation in 2D,
where H z is the unknown z-component of the magnetic field, ε r and μ r are relative permittivity and permeability values, respectively, and \( k_0 = {{2\pi } \mathord{\left/ {\vphantom {{2\pi } \lambda }} \right. } \lambda } \) is the incident wavevector magnitude. After solving Eq. 1, the in-plane components of \( \vec{E} \) were inferred from
For all calculations, circular-shaped computational domains with a 300 nm radius were used with the scattering object (Au nanoparticle dimer) modeled at the origin. NETGEN was used for domain discretization using triangular elements [28]. Within each element, H z was approximated using linear nodal basis functions,
where \( \phi_j^e \) is the value of H z at node j and \( N_j^e \) is a linear function defined only within the element e, which decreases linearly from 1 at node j to 0 at the face opposite to node j,
where \( a_j^e \), \( b_j^e \), and \( c_j^e \) are coefficients that depend on the geometry of e [17]. To simulate an open-region, the Sommerfield radiation condition was enforced on the exterior of the computational domain,
where \( r = \sqrt {x^2 + y^2} \).
Cross sections were calculated using the following integral expressions [29],
where \( \varepsilon_i \left( r \right) \) is the imaginary part of the relative permittivity, \( \vec{E}_0 \left( r \right) \) is the incident electric field, and * denotes complex conjugation.
Permittivity values of Au were calculated using a Drude plus 2 Lorentz pole dielectric model [30] fit to the empirically determined dielectric data of Lynch and Hunter [31] over the wavelengths important to this study (λ = 300–800 nm; we note that the empirical data could have been used directly; however, the difference between the two is negligible).
Results and discussion
TEM images of the SERS nanotags were obtained in order to determine realistic system parameters for the model nanoparticle dimer structure. Figure 1a demonstrates that the sample is predominately composed of aggregated particles containing approximately two to five nanoparticles, each with an average diameter of 90 nm and protective SiO2 shell with width of 50 nm. The inset in Fig. 1(a) shows a representative dimer structure where the nanoparticles are fused. The existence of dimers, where the interparticle distance is close to or less than zero highlights the need for robust computational methods to determine the effect of sub-nanometer features on EM properties (e.g., extinction spectra and EM enhancements). A representative image of Rayleigh scattering from the sample is presented in Fig. 1(b), with corresponding LSPR spectra of two individual particles shown in Figs. 1(c) and (d). The LSPR spectra of these individual diffraction-limited spots demonstrates that particles in the sample give rise to varied scattering spectra, consistent with a distribution of nanoparticle geometries and aggregations. In particular, the spectrum containing a single main peak is indicative of an individual nanoparticle, where a single dipole resonance is expected [32]. The other LSPR spectrum contains multiple peaks to the red of 500 nm, suggesting that the particle is a group of aggregated or very closely spaced nanoparticles (for aggregated nanoparticles there is a red-shift of the main dipole resonance relative to the monomer result near 600 nm, and peaks corresponding to higher-order multipole resonances, related to different distributions of polarization charge, are often observed [23]). Although the LSPR spectra can be used to infer nanoparticle structure, correlated high resolution-TEM (HR-TEM) and single-particle LSPR measurements are required to draw definitive conclusions [33]. Therefore, in order to determine how nanoparticle structure impacts the EM properties of specific nanostructures, such correlated HR-TEM and LSPR measurements are currently underway.

a TEM image of SiO2-coated Au nanoparticles showing the sample is predominately aggregates, (inset) fused nanoparticle dimer, b Rayleigh scattering from the nanoparticles deposited on glass, c–d corresponding LSPR spectra for particles 1 and 2 in b, respectively
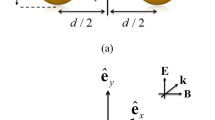
For our FEM calculations, we used the structural information from experiment and focused on a simple dimer structure with the nanoparticles arranged head to head, Fig. 1(a) inset. The structure was simplified by treating it as a 2D (nanowire) system. The use of a 2D rather than 3D model makes it possible to converge the EM field calculations with greater accuracy, which is important for the very small (less than 1 nm) separations that we consider. This simplification should not strongly influence the plasmon resonance spectrum (although, a slight blue-shift may occur because 2D resonances are often blue-shifted compared to 3D). A schematic diagram of the system under consideration is shown in Fig. 2: two 90-nm diameter infinite Au cylinders, each with a 20-nm thick SiO2 shell (which behaves similarly to a 50-nm shell), are separated by a distance d and illuminated using light polarized along the dimer axis.

Schematic diagram of Au cylinder dimer system modeled using FEM. The dotted line indicates the region where probability distributions, \( P\left( {\left| {\vec{E}} \right|^4 } \right) \), were calculated
Figure 3 presents extinction spectra for spacings of d = 5 to −10 nm (negative distances correspond to fused nanoparticles). The d = 5 nm separation spectrum is fairly close to what is found for a Au monomer, a single strong resonance near 600 nm. This result agrees qualitatively well with the experimental LSPR spectrum in Fig. 1(c), confirming the assumption that it corresponds to a relatively isolated nanoparticle. When d is reduced to 1 nm, the dipole resonance red-shifts to around 700 nm, and higher-order multipole resonances begin to appear near 600 nm, giving results that are similar to Fig. 1d. These results are consistent with those found by Kottman and Martin for coupled Ag nanowires [23]. The red-shifting increases as d decreases; and curiously, the fused structure with d = −1 nm is quite similar to that with d = 0.25 nm. For d values below −1 nm, the dipole resonance blue-shifts, eventually falling below 700 nm for d = −10 nm, consistent with the structure becomes less prolate.

Extinction cross sections for nanoparticle separations of d = 5 to −10 nm
Figure 4 presents the maximum EM enhancements (taken to be \( \left| {\vec{E}} \right|^4 \)), regardless of the position (although, below we show that this always occurs at the same spot), for the d under consideration. Broader peaks are seen than in Fig. 3, along with a sharp rise in EM enhancement as d is decreased. In particular, significant changes are observed as d is decreased from 1 to 0.5 to 0.25 nm, where the maximum EM enhancement increases by 3 orders of magnitude, from approximately 108 to 109 to 1010. Notice that d = 0.5 nm separation is needed to get EM enhancements above 108, a value considered to be necessary to give SMSERS.

Maximum EM enhancements \( \left( {\left| {\vec{E}} \right|^4 } \right) \) for nanoparticle separations of d = 5 to −10 nm
For all separations, the EM enhancement peaks occur near the multipole resonances in the extinction spectra, Fig. 3. In addition, the dipolar resonance (most red) was always found to be the most intense, consistent with results previously demonstrated by Hao and Schatz [19]. Furthermore, regardless of the wavelength, the maximum EM enhancement was always located at the junction region (along the dipolar axis). Considering that the higher-order multipole resonances are not oriented along this axis, it is surprising that even in these cases this is where the maximum EM enhancements were found (in these situations it is possible that hybrid high-order multipole–dipole resonances can occur [34]). However, this effect is understandable considering that the nanoparticle dimer acts as an antenna, concentrating EM fields at the junction. Figure 5a shows \( \left| {\vec{E}} \right|^4 \) at λ = 785 nm for d = 0.25 nm, demonstrating the extent of the localization of the EM enhancement (approximately 4 × 0.25 nm).

EM enhancement \( \left( {\left| {\vec{E}} \right|^4 } \right) \) at λ = 785 nm for nanoparticle separations of a d = 0.25 and b d = −1 nm
As soon as the nanoparticles fuse (d < 0), an extremely large increase in EM enhancements (over 4 orders of magnitude compared to d = 0.25 nm) to greater than 1014 and an extreme red-shift of the dipolar resonance to the red of 1,000 nm is observed. These EM enhancements are significantly larger than has been reported in most of the past EM studies of fused structures [6, 23, 35], and in fact they suggest that in these systems non-resonant SMSERS would be possible. The reason for this exotic behavior is that the crevice formed by the dimer overlap is incredibly sharp, yet the overlap is not severe enough to exclude the antenna position where strong localization of the EM enhancements occur. Figure 5b shows \( \left| {\vec{E}} \right|^4 \) at λ = 785 nm for d = −1 nm, where it is seen that the large EM enhancements come at the expense of the field being further localized to an area less than 0.1 × 0.1 nm, consistent with that previously found for arrays of fused cylinder structures [35]. As the dimer overlap is further increased, the EM enhancements rapidly decrease.
To correlate the EM enhancements with a SERS signal, we assumed that molecules with a diameter d m form a close packed monolayer around both nanoparticles (as long as they can fit). Probabilities of finding \( \left| {\vec{E}} \right|^4 \) at 1/2 of d m from the nanoparticle surface, \( P\left( {\left| {\vec{E}} \right|^4 } \right) \) (shown as a dotted line in Fig. 2), were calculated, Fig. 6. The purpose of this was twofold: one, to take into account that even though some structures give orders of magnitude greater EM enhancements than others (e.g., d = −1 nm), these are often highly confined (see Fig. 5); and two, to take into consideration that large molecules will not be able to fit into too small crevices and junctions where the EM enhancements are the most intense. Three molecular diameters were studied, d m = 0.25, 1.0, and 2.0 nm, for two nanoparticle separations, d = 0.25 and d = −1 nm (in Supporting Information we present results for nanoparticle separations of d = 5 to −20 nm). For all cases, almost 100% of \( P\left( {\left| {\vec{E}} \right|^4 } \right) \) is for \( \left| {\vec{E}} \right|^4 \), indicating that over the majority of the structure there is essentially no EM enhancement, which is confirmed by comparison with Fig. 5. A gradual decline in \( P\left( {\left| {\vec{E}} \right|^4 } \right) \) with increasing \( \left| {\vec{E}} \right|^4 \) is observed, indicating that higher EM enhancements are more strongly localized (however, it should be pointed out that for d m = 0.25 nm there is a strong drop in \( P\left( {\left| {\vec{E}} \right|^4 } \right) \) near \( \left| {\vec{E}} \right|^4 = 10^8 \), which then increases with \( \left| {\vec{E}} \right|^4 \). By comparison with Fig. 5, this is seen to occur because \( \left| {\vec{E}} \right|^4 \) decays significantly to the right of the junction region, a “shadowing” effect arising from the close nanoparticle spacing). The molecular size effects are apparent in Fig. 6, where it is seen that the larger the molecule the lower the maximum value of \( \left| {\vec{E}} \right|^4 \). In addition, for all cases except when d m = 0.25 nm and d = 0.25 nm, a sharp drop is seen in \( P\left( {\left| {\vec{E}} \right|^4 } \right) \) for the highest \( \left| {\vec{E}} \right|^4 \) values, indicating that the molecules cannot sample the entire maximum \( \left| {\vec{E}} \right|^4 \) region.

Probability distributions of \( \left| {\vec{E}} \right|^4 \), \( P\left( {\left| {\vec{E}} \right|^4 } \right) \), for nanoparticle separations of a d = 0.25 and b d = −1 nm
Two ways of correlating the EM enhancement results with the SERS signal were used. We first multiplied \( P\left( {\left| {\vec{E}} \right|^4 } \right) \) by \( \left| {\vec{E}} \right|^4 \), giving the expected signal distribution as a function of \( \left| {\vec{E}} \right|^4 \), Fig. 6. For d = 0.25 nm, the highest values of \( \left| {\vec{E}} \right|^4 \) are seen to be dominant. This is not surprising considering that for positive separations all of the molecules can sample locations with the highest EM enhancements. For d = −1 nm, the next to highest values are seen to be dominant. This result is also expected as the sharp crevices exclude molecules of any size entirely from the maximum \( \left| {\vec{E}} \right|^4 \)region. Next, we added up \( \left| {\vec{E}} \right|^4 \) at each position to give the average SERS signal. By then sorting the \( \left| {\vec{E}} \right|^4 \) values, we determined the percentage and number of molecules that contribute to a certain percentage of the signal, Fig. 7. For d = −1 nm spacing, less molecules are predicted to contribute to more of the signal compared to d = 0.25 nm spacing. Interestingly, for all d and d m, less than 3% of the molecules (a total of approximately 5–15) contribute to greater than 90% of the SERS signal. These results demonstrate a significant challenge for observing SMSERS, as for moderate concentrations a few molecules will always give the majority of the signal, and for low concentrations single molecules must find (and be able to fit in) highly localized positions of EM enhancement.

Percent (top) and corresponding number (bottom) of molecules that contribute to the SERS signal for nanoparticle separations of a d = 0.25 and b d = −1 nm
Conclusion
The FEM calculations show that the EM contribution to SERS in junction structures can completely dominate the signal, and in favorable cases enable the possibility of single-molecule SERS (SMSERS). While aspects of these conclusions match what has been found in earlier work, the greater accuracy in our FEM calculations has enabled the determination of new aspects of EM enhancements, such as extremely high (1014) enhancements for crevices that occur in fused structures. In addition, we have systematically determined intensity probability distribution information for molecules of various sizes on the nanoparticles, showing that smaller molecules can in some cases exhibit larger enhancement factors due to the high localization of the EM enhancements.
The results presented show some of the challenges associated with observing SMSERS, as the significant EM enhancements are highly localized and (assuming a moderate concentration) a few molecules are always responsible for most of the signal.
The comparison with experiment was provided for transmission electron microscopy (TEM) and localized surface plasmon resonance (LSPR) measurements, and the results suggest that dimer (and other structures) show plasmon excitations at wavelengths of 700 nm and longer, which we found correlate with the strongest SERS intensities. This provides important guidance for future work in which correlated TEM-LSPR-SERS measurements will be performed to assess the EM enhancement characteristics of a variety of structures. Further theory work will be aimed at providing a more quantitative assessment of the crevice results that were reported in this work. The importance of nonlocal dielectric effects also needs to be determined. In addition, it will be useful to study other fused nanoparticle structures to determine those that optimize the crevice contributions to SERS.
References
Albrecht MG, Creighton JA (1977) J Am Chem Soc 99:5215
Fleischman M, Hendra PJ, McQuillan AJ (1974) Chem Phys Lett 26:163
Jeanmaire DL, Van Duyne RP (1977) J Electroanal Chem 1:84
Schatz GC (1984) Acc Chem Res 17:370
Metiu H, Das P (1984) Annu Rev Phys Chem 35:507
Xu H, Aizpurua J, Kall M, Apell P (2000) Phys Rev E 62:4318
Schatz GC, Van Duyne RP (2002) In: Chalmers JM, Griffiths PR (eds) Handbook of vibrational spectroscopy. Wiley, New York
Nie S, Emory SR (1997) Science 275:1102
Kneipp K, Wang Y, Kneipp H, Perelman LT, Itzkan I, Dasari RR, Feld MS (1997) Phys Rev Lett 78:1667
Dieringer JA, Wustholz KL, Masiello DJ, Camden JP, Kleinman SL, Schatz GC, Van Duyne RP (2009) J Am Chem Soc 131:849
Kerker M, Wang DS, Chew H (1980) Appl Opt 19:3373
Doering WE, Piotti ME, Natan MJ, Freeman RG (2007) Adv Mater 19:3100
Draine BT, Flatau PJ (1994) J Opt Soc Am A 11:1491
Draine BT, Flatau PJ (2003) User guide for the discrete dipole approximation code DDSCAT.6.0. Available from: <http://arxiv.org/ags.astro-ph/0309069>
Taflove A, Hagness SC (2005) Computational electrodynamics: the finite-difference time-domain method, 3rd edn. Artech House, Inc., Norwood
Yee SK (1966) IEEE Trans Antennas Propagat 14:302
Jin J (2002) The finite element method in electromagnetics, 2nd edn. Wiley, New York
Kottmann JP, Martin OJF, Smith DR, Schultz S (2000) Opt Express 6:213
Hao E, Schatz GC (2004) J Chem Phys 120:357
Zou S, Schatz GC (2005) Chem Phys Lett 403:62
Xu H (2004) Appl Phys Lett 85:5980
Zeman EJ, Carron KT, Schatz GC, Van Duyne RP (1987) J Chem Phys 87:4189
Kottmann JP, Martin OJF (2001) Opt Express 8:655
Aravind PK, Nitzan A, Metiu H (1981) Surf Sci 110:189
Vanin AI (1995) J Appl Spect 62
Felidj N, Aubard J, Levi G (1999) J Chem Phys 111:1195
McMahon JM (2009) JFEM2D. Available from: <http://www.thecomputationalphysicist.com>
Schöberl J (2009) NETGEN. Available from <http://www.hpfem.jku.at/netgen/>
Saxon DS (1955) UCLA Department of Meteorological Science Report 9
McMahon JM, Gray SK, Schatz GC (2009) Surface nanophotonics theory. In: Wiederrecht G (ed) Comprehensive nanoscience and technology. Elsevier, Amsterdam
Lynch DW, Hunter WR (1985) In: Palik ED (ed) Handbook of optical constants of solids. Academic, Orlando
Bohren CF, Huffman DR (1983) Absorption and scattering of light by small particles. Wiley, New York
McMahon JM, Wang Y, Sherry LJ, Van Duyne RP, Marks LD, Gray SK, Schatz GC (2009) J Phys Chem C 113:2731–2735
Prodan E, Radloff C, Halas NJ, Nordlander P (2003) Science 302:419
Garcia-Vidal FJ, Pendry JB (1996) Phys Rev Lett 77:1163
Acknowledgment
This research was supported by the NSF (CHE-0414554), AFOSR/DARPA Project BAA07-61 (FA9550-08-1-0221), and NSF MRSEC (DMR-0520513) at the Materials Research Center of Northwestern University. We thank the NUANCE Center at Northwestern University for providing access to the TEM equipment.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
Probability distributions of electromagnetic enhancements for nanoparticle separations from 5 to −20 nm; Percentage and number of molecules that contribute to the SERS signal for particle separations of 5 to −20 nm and molecular diameters of 0.25 to 2 nm (PDF 195 kb).
Rights and permissions
About this article
Cite this article
McMahon, J.M., Henry, AI., Wustholz, K.L. et al. Gold nanoparticle dimer plasmonics: finite element method calculations of the electromagnetic enhancement to surface-enhanced Raman spectroscopy. Anal Bioanal Chem 394, 1819–1825 (2009). https://doi.org/10.1007/s00216-009-2738-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-2738-4