
Abstract
The metabolism of the selective estrogen receptor modulator toremifene was simulated in an on-line electrochemistry/enzyme reactor/liquid chromatography/mass spectrometry system. To simulate the oxidative phase I metabolism, toremifene was oxidized in an electrochemical (EC) flow-through cell at 1,500 mV vs. Pd/H2 to its phase I metabolites, some of which are reactive quinoid species. In the presence of glutathione-S-transferase (GST), these quinoid compounds react with glutathione, which is also the common detoxification mechanism in the body. While reacting with glutathione, the chlorine atom is eliminated from the toremifene moiety. Due to higher conversion rates, GST supplied in continuous flow proved to be more efficient than using immobilized GST on magnetic microparticles. In the absence of GST, not all GSH adducts are formed, proving the necessity of a phase II enzyme to simulate the complete metabolic pathway of xenobiotics in an on-line EC/LC/MS system.

Mass voltammogram of toremifene
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the development process of a new chemical entity, metabolism studies are indispensable [1–6]. The metabolism of xenobiotics such as drugs or environmental compounds is generally divided into two different phases [7, 8]. Phase I reactions are mainly oxidation reactions, catalyzed by enzymes from the cytochrome P450 family in the liver [9, 10]. The resulting phase I metabolite may either be excreted directly by the kidneys or the bile, or it can undergo a phase II metabolism step, in which small and hydrophilic biomolecules, such as glutathione (GSH) or glucuronic acid, are coupled to the phase I metabolite. This process is catalyzed by an appropriate transferase [11]. Besides common in vitro systems using hepatocytes or microsomes [12–14] and in vivo methods, in which radioactively labeled drug compounds are administered [15, 16], there is an increasing interest in new simulation methods providing rapid and reliable information on possible metabolites. Applying these methods in an early stage of metabolic studies may lead to a more efficient and economic procedure in drug development. Besides metabolism simulation in silico [17, 18], the chemical simulation using appropriate metal-porphyrin complexes that mimic the active center of a metabolic enzyme [19–22] and the electrochemical (EC) simulation of oxidative phase I reactions are of considerable interest. The advantage of such simulation methods is that the complexity of the system is reduced to the basic factors so that the simulation is relatively easy to conduct. Particularly in an on-line combination with an LC/MS system, the EC simulation features some interesting aspects and has proven its value in recent years [23–36]. Both the generation and identification of metabolites takes place in the same system, thus saving time, effort, and costs. Furthermore, information about oxidatively labile sites in a molecule is easily accessible. However, only insufficient attention was paid so far to the systematic simulation of conjugative phase II metabolism reactions, which are inherent parts of the complete biotransformation process.
To investigate the potential of on-line EC/LC/MS for the additional simulation of conjugative phase II reactions, the selective estrogen receptor modulator toremifene was selected as model compound (Fig. 1). Toremifene, as well as the better-known analogous tamoxifene, were used for the treatment of breast cancer [37, 38]. The cytochrome P450 catalyzed metabolism of toremifene and tamoxifene in the liver comprises mainly hydroxylation, N-desalkylation, N-oxide formation. Furthermore, after either hydroxylation or O-desalkylation, dehydrogenation to reactive quinone methides may occur [39–42]. The reactive quinoid phase I metabolites are detoxified by reaction with glutathione (GSH) under catalysis of glutathione-S-transferase (GST). In case of tamoxifene, a 1,4-Michael addition of GSH to the quinone methide takes place, while in the toremifene quinone methide, chlorine is nucleophilically substituted by GSH. Although reactions between electrochemically generated reactive phase I metabolites and GSH have been reported [25, 28–30, 33–35], none of these reactions included a nucleophilic substitution of a halogen atom, suggesting that this reaction requires enzymatic catalysis to proceed. Therefore, toremifene is a good example to study an enzymatic phase II reaction embedded in an EC/LC/MS system. The respective investigations are presented in this work.

Structure of the selective estrogen receptor modulator toremifene
Experimental section
Chemicals and materials
Toremifene was obtained from LKT Laboratories Inc. (St. Paul, MN, USA), reduced L-glutathione, glutathione-S-transferase from equine liver, ammonium formate, 2-(N-morpholino)ethanesulfonic acid monohydrate (MES), sodium carbonate, glycine- and phosphate-buffered saline tablets were purchased from Sigma-Aldrich Chemie GmbH (Steinheim, Germany). Ammonium hydroxide solution and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) were obtained from Fluka Chemie GmbH (Buchs, Switzerland). Acetonitrile (HPLC grade) and N-hydroxysuccinimide were ordered from Merck KGaA (Darmstadt, Germany). All chemicals were used in the highest quality available. Water used for HPLC was purified using a Milli-Q Gradient A 10 system and filtered through a 0.22 µm Millipak 40 filter (Millipore, Billerica, MA, USA). Magnetic latex microparticles (type: micromer-M®) with 3 µm diameter and carboxylic acid functionalization were ordered from micromod Partikeltechnologie GmbH (Rostock, Germany). Permanent magnets were purchased via www.supermagnete.de (Webcraft GmbH, Uster, Switzerland).
Instrumentation
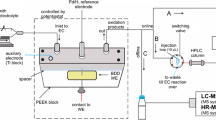
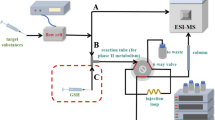
The electrochemical oxidation was performed using a model 5021 conditioning cell (ESA Biosciences Inc., Chelmsford, MA, USA), which was controlled by a home-made potentiostat. The EC cell contained a porous glassy carbon working electrode with a large surface area, a Pd counter electrode and a Pd/H2 reference electrode. A PEEK in-line filter was placed in front of the cell inlet to protect the working electrode. For the EC/MS measurements, a micrOTOF mass spectrometer (Bruker Daltonics, Bremen, Germany), equipped with an electrospray ionization source was used. The software used for controlling the micrOTOF and data processing was micrOTOF control 1.1 and DataAnalysis 3.3 (Bruker Daltonics). For EC/enzyme/LC/MS measurements, a Shimadzu HPLC system (Duisburg, Germany) comprising two LC-10ADvp pumps, a DGC-14A degasser, a SIL-HTa autosampler, a CTO-10Avp column oven and a SPD-10AVvp UV detector was coupled to a QTRAP mass spectrometer from Applied Biosystems (Darmstadt, Germany) with an electrospray ionization (ESI) source. The software used for controlling HPLC and QTRAP was Analyst 1.4.1 (Applied Biosystems).
Immobilization of GST
A 0.5 mL micromer-M® suspension (surface functionalization: COOH, c = 50 mg/mL) was transferred into an Eppendorf tube. The particles were separated using a permanent magnet and the supernatant was removed. The particles were resuspended in 1 mL aqueous 0.1 M MES buffer (pH adjusted to 6.3 with aqueous 0.5 M Na2CO3 solution). The particles were again separated at a permanent magnet and the supernatant was removed. 4 mg N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and 8 mg N-hydroxysuccinimide were dissolved in 1 mL 0.1 M MES buffer (pH 6.3) and the solution was added to the particles. After vortexing for 1 min, the suspension was incubated for 2 h at room temperature under continuous mixing. After the incubation, the activated particles were separated by a permanent magnet and washed with 1 mL 0.1 M MES buffer (pH 6.3). The activated particles were resuspended in 1 mL PBS buffer (pH 7.4) containing 200 µg GST. The suspension was incubated overnight at room temperature under continuous mixing before the particles were separated at a permanent magnet. For blocking activated reaction sites at the surface of the particles, 1 mL of a 25 mM solution of glycine in PBS buffer (pH 7.4) was added, and the suspension was incubated for 30 min at room temperature under continuous mixing. The functionalized particles were washed twice with 1 mL PBS buffer (pH 7.4) and used immediately after resuspension in 1 mL PBS buffer.
EC oxidation and enzymatic phase II reaction
For the electrochemical oxidation, a 100 µM solution of toremifene in ammonium formate solution (20 mM, pH adjusted to 7.4 with ammonium hydroxide solution) and methanol (50/50, v/v) was pumped through the EC cell at a flow rate of 2 µL/min. For direct infusion of the electrochemical oxidation mixture, the effluent of the EC cell was directly conducted into the ESI source of the micrOTOF mass spectrometer. In these experiments, the distance between EC cell and MS was about 50 cm, corresponding to approximately 6 µL void volume. No disturbing electrical currents from the ESI source were observed at the EC cell. The potential at the working electrode of the EC cell was controlled using a home-made potentiostat. The phase II metabolism was initiated either by adding GST in continuous flow or by using GST, which was immobilized on magnetic microparticles. For the continuous-flow set-up, a solution of GSH (500 µM) and GST (50 µg/mL) in 20 mM ammonium formate (pH 7.4) was added via a T-piece at a flow rate of 4 µL/min. When using the magnetic particles, only GSH (500 µM) was added at the same flow rate via a T-piece. In both cases, the complete flow was pumped through a reaction tube with a length of 160 mm and an inner diameter of 0.762 mm. In the continuous-flow set-up, this reaction coil was empty, while in case of the immobilization set-up, the magnetic particles, functionalized with GST, were filled into the coil and fixed with two permanent magnets at the outer wall of the tube over a length of 100 mm. Additionally, a PEEK in-line filter was placed at the outlet of the cell to prevent the magnetic microparticles from being flushed into the LC/MS system. In both set-ups, the effluent from the reaction coil was collected in a 10 μL injection loop that was mounted on a ten-port switching valve. After switching the valve, the content of the loop was emptied onto the HPLC column [31].
HPLC conditions
The separation of toremifene, its oxidation products and GSH adducts was performed on a ZORBAX Eclipse XDB-C8 column (150 × 2.1 mm, 3.5 µm particle size, Agilent, Waldbronn, Germany) at 40°C and a flow rate of the mobile phase of 250 µL/min. Eluent A of the mobile phase was an aqueous solution of 20 mM ammonium formate (pH adjusted to 7.4 with ammonium hydroxide solution) and eluent B was methanol. The injection volume was 10 μL (loop size). The gradient profile for the separation is shown in Table 1.
MS conditions
On the micrOTOF instrument, full scan spectra (m/z 150–1,000) were recorded after direct infusion using ESI(+)-MS under the following conditions: end plate offset: −200 V, capillary: −4,500 V, nebulizer gas (N2), 0.8 bar, drying gas (N2), 4.0 L/min, drying temperature, 180°C, capillary exit, 120.0 V, skimmer 1, 40.0 V, skimmer 2, 23.0 V, hexapole 1, 20.0 V, hexapole 2, 20.6 V, hexapole RF, 150 V, transfer time, 49.0 μs, pre-pulse storage, 5.0 μs, detector: −1,000 V. Internal calibration was performed using sodium formate clusters before each analysis. On the QTRAP instrument, ESI(+)-MS was performed using the linear ion trap in enhanced mode. The following settings were used: nebulizer gas, 50 psi, dry gas/heating gas, 70 psi, curtain gas, 25 psi, temperature, 400°C, ionspray voltage, 5,500 V, declustering potential, 20 V, entrance potential, 7 V, collision energy, 10 V, CAD gas pressure: “high”.
Results and discussion
Electrochemical oxidation of toremifene
The selective estrogen receptor modulator toremifene (Fig. 1) was selected as model compound to investigate the potential of the electrochemistry/immobilized enzymes/liquid chromatography/mass spectrometry system since the phase I and II metabolism of this and analogous compounds has been well investigated [39–42]. To obtain first information on the electrochemical oxidation behavior of toremifene, the set-up was simplified to only electrochemistry/mass spectrometry. Toremifene was pumped continuously through the EC cell and the outlet of the cell was directly connected to the ESI source of a time-of-flight mass spectrometer. The potential at the working electrode of the EC cell was ramped between 0 and 2,000 mV vs. Pd/H2 in 20 min (100 mV/min). The mass spectra of the oxidation mixture were recorded in dependence of the EC potential, which is shown in Fig. 2. From the resulting “mass voltammogram”, rapid insight into the oxidation behavior of toremifene can be obtained.

Mass spectra of the oxidation products of toremifene in dependence of the oxidation potential (“mass voltammogram”). The ramping velocity was 100 mV/min
The oxidation of toremifene (m/z 406.19) starts in the range of 700 mV vs. Pd/H2, when a compound with m/z 392.18 is formed. This corresponds to a formal loss of CH2, resulting from the elimination of one N-methyl group. Further oxidation yields also the bis-demethylated product, which was observed at m/z 378.16. The formation of this bis-demethylated toremifene starts at about 900 mV vs. Pd/H2. The formation of N-demethylated metabolites is also observed under cytochrome P450 catalysis of toremifene in rat, mouse, and human liver microsomes [42], demonstrating the good accordance between EC simulation and in vitro experiments. Besides the demethylated products, the oxidation of toremifene, combined with the gain of one or more oxygen atoms, could be observed as well. For example, a species with m/z 422.18 was detected, which is presumably hydroxylated toremifene. Another oxidation reaction yields dehydrogenated toremifene with m/z 404.18. The most interesting oxidation product with an m/z ratio of 333.10 is formed at 1,000 mV vs. Pd/H2. The exact mass refers to a sum formula of C22H18OCl for the [M + H]+, which corresponds to an O-desalkylation aligned with a dehydrogenation under loss of two hydrogen atoms, yielding a quinone methide. The possible reaction pathway of the O-desalkylation and dehydrogenation at 1,500 mV vs. Pd/H2 is presented in Fig. 3.

Oxidation scheme of toremifene at 1,500 mV vs. Pd/H2
The formation of the toremifene quinone methide is of particular interest, since the resulting metabolite is a quinoid species of which similar metabolites have been previously described, mostly after hydroxylation at one of the unsubstituted aromatic rings [39, 40]. Interestingly, for tamoxifene, which differs from toremifene only in the missing chlorine atom, a quinone methide, formed after O-desalkylation, has been reported [41] and is called “metabolite E”. “Metabolite E”, as well as similar quinoid species of toremifene, is detoxified in common in vitro and in vivo assays in a phase II reaction step by a nucleophilic attack of GSH. In case of tamoxifene, a 1,4-Michael addition yields a common GSH adduct, which is consistent with the conjugation products of similar quinoid species (e.g., NAPQI, [25, 29]). The most electrophilic site in the toremifene quinone methide molecule is the chlorinated C atom, which is indeed the location where in most cases GSH nucleophilically attacks the toremifene quinone methide [39–41]. Upon this reaction, the chlorine atom is substituted by a GSH residue and GSH adducts are formed via a nucleophilic substitution. While 1,4-Michael addition reactions with GSH often proceed spontaneously after electrochemical oxidation of drug compounds in the presence of GSH [22, 28, 30, 31, 33–35], nucleophilic substitution of a halogen atom after electrochemical oxidation has not been observed so far. Therefore, it is of particular interest if such substitution reactions proceed under enzyme catalysis, which should be investigated with toremifene.
EC in combination with enzymatic phase II reaction and LC/MS
To simulate both phase I and phase II metabolism of toremifene in an on-line system, the enzyme catalyzed conjugative phase II metabolism step has to be embedded into the EC/LC/MS system. Therefore, glutathione-S-transferase, which is the respective enzyme catalyzing the nucleophilic attack of GSH to phase I products, was implemented in the set-up. A constant EC potential of 1,500 mV vs. Pd/H2 was selected, since the yield of the particular quinone methide metabolite was maximized at this potential. The mass spectrum of toremifene after oxidation at 1500 mV vs. Pd/H2 is shown in Fig. 4. At this potential, both the mono-demethylated toremifene and the bis-demethylated metabolite are detected. Both compounds underwent an additional dehydrogenation reaction to the species with m/z 390.16 and m/z 376.16, respectively. The hydroxylated toremifene metabolites are mostly dehydrogenated as well. The O-desalkylated metabolite shows good signal intensity.

Mass spectrum of the oxidation products of toremifene at 1,500 mV vs. Pd/H2
To initiate the enzyme catalyzed reaction, the effluent from the EC cell was combined with a stream from a second syringe pump via a T-piece (Fig. 5). The combined streams were conducted through a reaction coil and the effluent from the reaction coil was collected in a 10 µL injection loop, which was mounted on a 10-port-switching valve [31]. The HPLC separation was started by switching the valve.

Schematic set-up of the EC/GST/LC/MS system for the simulation of the oxidative phase I and the conjugative phase II metabolism of toremifene
In the first experiment, the second syringe contained only aqueous buffer (pH 7.4). The reason was to optimize the HPLC separation in respect of the oxidation products. Figure 6a shows the HPLC chromatogram for the extracted ion trace with m/z 333.10, corresponding to the O-desalkylated toremifene quinone methide. Interestingly, besides a large peak with a retention time of 19.3 min, a smaller peak with a slightly higher retention time and the same m/z could be detected. Apparently, two isomers with the same m/z are formed, which, however, could not be further identified.

Combined extracted ion traces of m/z 333.10 (toremifene quinone methide) and m/z 604.21 (GSH adduct) of the HPLC separation of toremifene a without addition of GSH; b with addition of GSH in absence of GST and c with addition of GSH in the presence of GST in continuous flow
In the next step, GSH in aqueous buffer (pH 7.4) was added via the second syringe. Yet, no enzyme for catalyzing the reaction with GSH was present. This experiment was used to identify possible GSH adducts that were formed spontaneously in absence of a catalyzing transferase. In fact, one GSH adduct was detected, having an m/z ratio of 604.21, which did not show a typical chlorine isotopic pattern. This mass was assigned to a sum formula of C32H34N3O7S for the [M + H]+, which formally corresponds to a substitution of the chlorine atom of the quinone methide by a GSH residue. The corresponding chromatogram with the extracted ion traces of the toremifene quinone methide and the GSH adduct with m/z 604.21 is shown in Fig. 6b. The GSH adduct elutes at 4.2 min as single peak. Furthermore, as can be seen from this chromatogram, the smaller peak with the higher retention time and with m/z 333.10 has disappeared. It thus seems obvious that this later eluting isomer of the quinone methide reacts already spontaneously with GSH under formation of a GSH adduct in which the chlorine atom is eliminated.
Eventually, GSH as well as GST were filled into the second syringe. In this set-up, GST can catalyze the reaction between GSH and electrochemically formed metabolites of toremifene. The chromatogram of the extracted ion traces with m/z 333.10 and 604.21 is shown in Fig. 6c. In this chromatogram, no quinone methide with m/z 333.10 could be detected anymore. Instead, a second peak with m/z 604.21 and a retention time of 7.8 min was observed. Hence, this compound is the reaction product, which is formed under GST catalysis of GSH and the toremifene quinone methide with a retention time of 19.3 min. However, the exact structure of this GSH adduct as well as of the one eluting at 4.2 min, which is already formed without enzyme catalysis could not be elucidated. By all means, the quinoid nature is lost after reaction with GSH as could be seen from the respective UV traces of the chromatograms, which should show a characteristic absorption in the range of 400 nm for quinoid species. Other GSH adducts were not detected, and bis-GSH adducts were only present to a minor extent.
The absence of GSH adducts of other oxidation products is interesting: If the oxidation products (as for example the dehydrogenated bis-demethylated compound with m/z 376.16, the dehydrogenated toremifene with m/z 404.18 and the dehydrogenated and hydroxylated compounds) were quinoid species, they would be expected to react readily with nucleophiles such as GSH. Since no other oxidation product besides the quinone methide with m/z 333.10 reacts with GSH, it is likely that these compounds are not quinoid species. Another hint supporting this assumption is the UV absorption of these compounds: quinoid species should show a strong absorbance at 400 nm in the UV trace of the chromatogram, which is, however, not the case for all dehydrogenated compounds except for the quinone methide itself with m/z 333.10. From the UV absorption as well as from the lack of the GSH adduct formation, a quinoid nature of the dehydrogenated bis-demethylated toremifene with m/z 376.16, the dehydrogenated toremifene with m/z 404.18 and the dehydrogenated hydroxylated compounds can be excluded. Since these compounds have two hydrogen atoms less than the respective derivatives with the toremifene structure, possibly a ring-closing reaction has occurred to fulfill the number of double bond equivalents. Such a ring-closing reaction might also have occurred upon the reaction of GSH and the quinone methides with m/z 333.10, which could explain the loss of the quinoid nature of the adducts.
In an alternative approach, GST was immobilized on magnetic microparticles, which were filled into the reaction tube and fixed with two permanent magnets outside of the tube. The advantage of using immobilized enzymes is the lower enzyme consumption. However, although the results were qualitatively in consistence with the continuous-flow experiments, the yield of the GSH adducts was considerably lower. Possible reasons for this might be an activity loss of GST during the immobilization procedure or that the amount of immobilized GST on the particles is too low for sufficient conversion. While the potential activity loss is a serious problem, the possibly too low amount of immobilized enzyme could be compensated by increasing the length of the reaction coil with the magnetic particles or improved by further optimization of the immobilization procedure.
However, the importance of the enzymatic phase II simulation in addition to the electrochemical phase I simulation is obvious, since without the addition of the phase II enzyme, the complete metabolism comprehending both the phase I and the phase II step with the formation of two isomeric GSH adducts could not be mimicked. Although previous approaches indeed include the reaction of electrochemically generated phase I metabolites with GSH [25, 28–30, 33, 34], in all cases, the reaction took already place in the absence of the respective phase II enzymes. However, the high reactivity of the phase I metabolites can not always be taken for granted, and for the complete simulation of the xenobiotic metabolism in an LC/MS system, the use of transferases will result in a more complete metabolism, as can be seen for the example of toremifene.
Conclusions
In summary, it can be emphasized that for the simulation of the complete spectrum of metabolism reactions, both the oxidative phase I metabolism and the conjugative phase II metabolism have to be considered separately. An enzymatically catalyzed phase II reaction was successfully implemented additionally to an electrochemical phase I simulation into an LC/MS system. Using this set-up, also phase II reactions that do not proceed without enzymes can be simulated on-line. In the future, the use of other phase II enzymes such as the important UDP-glucuronosyltransferase is an interesting application of the on-line system since the reaction of glucuronic acid with hydroxyl groups of phase I metabolites is another common phase II step, but does not occur without enzymatic catalysis.
References
Grossman SJ (2008) Overview: drug metabolism in the modern pharmaceutical industry. In: Zhang D, Zhu M, Humphreys WG (eds) Drug metabolism in drug design and development. Wiley, Hoboken
Humphreys WG, Unger SE (2006) Chem Res Toxicol 19:1564
Davis-Bruno KL, Atrakchi A (2006) Chem Res Toxicol 19:1561
Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA (2004) Chem Res Toxicol 17:3
Guengerich FP (2006) Chem Res Toxicol 19:1559
Guengerich FP, MacDonald JS (2007) Chem Res Toxicol 20:344
Iyer KR, Sinz MW (1999) Chem Biol Interact 118:151
Josephy PD, Guengerich FP, Miners JO (2005) Drug Metab Rev 37:575
Lamb DC, Waterman MR, Kelly SL, Guengerich FP (2007) Curr Opin Biotechnol 18:504
Guengerich FP (2008) Chem Res Toxicol 21:70
Remmel R, Nagar S, Argikar U (2008) Conjugative metabolism of drugs. In: Zhang D, Zhu M, Humphreys WG (eds) Drug metabolism in drug design and development. Wiley, Hoboken
Brandon EFA, Raap CD, Meijerman I, Beijnen JH, Schellens JHM (2003) Toxicol Appl Pharmacol 189:233
Pelkonen O, Turpeinen M, Uusitalo J, Rautio A, Raunio H (2005) Basic Clin Pharmacol Toxicol 96:167
Plant N (2004) Drug Discovery Today 9:328
Marathe PH, Shyu WC, Humphreys WG (2004) Curr Pharm Des 10:2991
Dalvie D (2000) Curr Pharm Des 6:1009
Kulkarni SA, Zhu J, Blechinger S (2005) Xenobiotica 25:955
Gombar VK, Alberts JJ, Cassidy KC, Mattioni BE, Mohutsky MA (2006) Curr Comput-Aided Drug Des 2:177
Meunier B, Bernadou J (2004) Adv Synth Catal 346:171
Feiters MC, Rowan AE, Nolte RJM (2000) Chem Soc Rev 29:375
Meunier B (1992) Chem Rev 92:1411
Lohmann W, Karst U (2008) Anal Bioanal Chem 391:79
Karst U (2004) Angew Chem Int Ed 43:2476
Permentier HP, Bruins AP, Bischoff R (2008) Mini-Rev Med Chem 8:46
Getek TA, Korfmacher WA, McRae TA, Hinson JA (1989) J Chromatogr A 474:245
Jurva U, Wikström HV, Bruins AP (2000) Rapid Commun Mass Spectrom 14:529
Jurva U, Wikström HV, Weidolf L, Bruins AP (2003) Rapid Commun Mass Spectrom 17:800
Van Leeuwen SM, Blankert B, Kauffmann JM, Karst U (2005) Anal Bioanal Chem 382:742
Lohmann W, Karst U (2006) Anal Bioanal Chem 386:1701
Madsen KG, Olsen J, Skonberg C, Hansen SH, Jurva (2007) Chem Res Toxicol 20:821
Lohmann W, Karst U (2007) Anal Chem 79:6831
Johansson T, Weidolf L, Jurva U (2007) Rapid Commun Mass Spectrom 21:2323
Jurva U, Holmen A, Groenberg G, Masimirembwa C, Weidolf L (2008) Chem Res Toxicol 21:928
Madsen KG, Skonberg C, Jurva U, Cornett C, Hansen SH, Johansen TH (2008) Chem Res Toxicol 21:1107
Lohmann W, Dötzer R, Gütter G, Van Leeuwen SM, Karst U (2008) J Am Soc Mass Spectrom doi:10.1016/j.jasms.2008.09.003
Thevis M, Lohmann W, Schrader Y, Kohler M, Bornatsch W, Karst U, Schänzer W (2008) Eur J Mass Spectrom 14:163
Valavaara R, Pyrrhonen S, Meikkinen M, Rissanan P, Blanco G, Tholix E, Nordmann E, Taskinen P, Holsti L, Hajba (1988) Eur J Cancer Clin Oncol 24:785
Williams GM, Ross PM, Jeffry AM, Karlsson S (1998) Drug Chem Toxicol 21:449
Fan PW, Zhang F, Bolton JL (2000) Chem Res Toxicol 13:45
Yao D, Zhang F, Yu L, Yang Y, van Breemen RB, Bolton JL (2001) Chem Res Toxicol 14:1643
Fan PW, Bolton JL (2001) Drug Metab Dispos 29:891
Jones RM, Lim CK (2002) Biomed Chromatogr 16:361
Acknowledgments
Financial support by the Deutsche Forschungsgemeinschaft (DFG, Bonn, Germany) and the Fonds der Chemischen Industrie (Frankfurt/Main, Germany) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lohmann, W., Karst, U. Electrochemistry meets enzymes: instrumental on-line simulation of oxidative and conjugative metabolism reactions of toremifene. Anal Bioanal Chem 394, 1341–1348 (2009). https://doi.org/10.1007/s00216-008-2586-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-2586-7