
Abstract
Artemisinin is an endoperoxide sesquiterpene lactone isolated from the Chinese medicinal plant Artemisia annua L. It has been widely used in South-East Asia and Africa as an effective drug against sensitive and multidrug-resistant Plasmodium falciparum malaria. A monoclonal antibody (mAb), designated as 3H2, was generated with artesunate–bovine serum albumin conjugate as the immunogen. mAb 3H2 was used to develop a highly sensitive and specific indirect competitive enzyme-linked immunosorbent assay (icELISA) for artemisinin. The concentration of analyte producing 50% of inhibition (IC50) and the working range of the icELISA were 1.3 and 0.2–5.8 ng/mL, respectively. The mAb 3H2 recognized the artemisinin analogs artesunate, dihydroartemisinin, and artemether with cross-reactivity of 650%, 57%, and 3%, respectively, but negligibly recognized deoxyartemisinin and the artemisinin precursors arteannuin B and artemisinic acid. The average recoveries of artemisinin fortified in A. annua samples at concentrations from 156 to 5,000 μg/g determined by icELISA ranged from 91% to 98%. The icELISA was applied for the determination of artemisinin in different wild A. annua samples and the results were confirmed by high-performance liquid chromatography (HPLC) analysis. The correlation coefficient (R 2) between the two assays was larger than 0.99, demonstrating a good agreement between the icELISA and HPLC results. This ELISA is suitable for quality assurance of A. annua L. materials.

Artemisia annua plant and antimalarial drugs derived from artemisinin
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Malaria is a global severe parasitic infection disease, particularly in South-East Asia and Africa. There are more than 500 million malaria cases annually. Due to the development of drug resistance to the conventional antimalaria drugs such as chloroquine and sulfadoxine–pyrimethamine, the World Health Organization now recommends treating uncomplicated malaria with artemisinin-based combination therapies. Artemisinin (Fig. 1), an endoperoxide sesquiterpene lactone isolated from the Chinese medicinal plant Artemisia annua L., is effective against sensitive and multidrug-resistant strains of the protozoan parasite Plasmodium falciparum that causes cerebral malaria [1–3]. Currently, A. annua is the main source of artemisinin since the complete organic synthesis is not economically feasible [4, 5], and the production of artemisinin from other biotechnological approaches is still on the way [6]. Therefore, high content of artemisinin in A. annua raw materials is essential for economical production of quality artemisinin to meet the needs, particularly in developing countries.

Structures of artemisinin and its analogs
In order to understand biosynthesis and metabolic pathways of artemisinin in plant and the pharmacokinetics in human body and to screen a large number of wild and cultured A. annua samples for quality control, a sensitive and specific analytical method for artemisinin is desirable. A number of methods have been developed for the detection of artemisinin, including high-performance liquid chromatography-UV detection (HPLC-UV) [7-10], HPLC-electrochemical detection (HPLC-ECD) [11–13], HPLC-evaporative light scattering detection [14–16], gas chromatography–flame ionization detection [16], GC–mass spectrometric detection [17–19], liquid chromatography–mass spectrometry [20], radioimmunoassay (RIA) [21], and enzyme-linked immunosorbent assay (ELISA) [22–25]. Among these methods, the instrumental methods are expensive and time-consuming and need rigorous sample preparation. RIA is more sensitive than the conventional methods, but it possesses shortcomings of health hazards and high radioactive waste disposal costs. In contrast, ELISA is rapid, cost-effective, selective, and sensitive and becomes popular for the detection of plant secondary metabolites [26, 27].
The published RIA and ELISA procedures [21–25] for the determination of artemisinin were based on either polyclonal antibodies (pAbs) or monoclonal antibodies (mAbs). Artemisinin content in the A. annua plants is very low, and other constituents in the plants would interfere with the determination of artemisinin. An immunoassay may meet the need if it is highly sensitive and specific. The published pAb-based assays [21–23] were not specific enough and cross-reacted with some artemisinin analogs present in A. annua samples, which resulted in the overestimation of artemisinin content in A. annua samples compared with HPLC-ECD [23]. The published mAb-based ELISAs [24, 25] were not sensitive enough to allow high dilution which would help to reduce the interference of other constituents in the A. annua plants, and the cross-reactivities of their mAbs with arteannuin B and artemisinic acid, of which the content was high in the A. annua plants, were not determined, so the accuracy of their mAb-based ELISAs used to quantify artemisinin in A. annua samples remains undetermined. We now report the production of a mAb against artemisinin with high sensitivity and specificity and the establishment of an indirect competitive ELISA (icELISA). The icELISA was applied to quantify artemisinin in crude extracts of wild A. annua from different Provinces in China, and the results were confirmed with HPLC.
Materials and methods
Materials
Artemisinin, artesunate, dihydroartemisinin, and artemether were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Arteannuin B, artemisinic acid, and deoxyartemisinin were kindly supplied by the China Academy of Chinese Medical Sciences (Beijing, China). Tri-n-butylamine, isobutylchloroformate, bovine serum albumin (BSA), ovalbumin (OVA), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride, polyethylene glycol 2000, cell freezing medium-dimethyl sulfoxide (DMSO; serum free), hypoxanthine, aminopterin, and thymidine (HAT), hypoxanthine and thymidine (HT) medium supplements, penicillin, streptomycin, l-glutamine, horse-radish-peroxidase-labeled goat antimouse IgG, complete and incomplete Freund’s adjuvant, and o-phenylenediamine (OPD) were purchased from Sigma (St Louis, MO, USA). Cell culture medium (Dulbecco’s modified Eagle’s medium, DMEM) and fetal bovine serum (FBS) were obtained from Gibco BRL (PaisLey, Scotland). Mouse antibody isotyping kit was obtained from Pierce (Rockford, IL, USA). All other chemicals and organic solvents used were of analytical grade.
Cell culture plates and 96-well polystyrene microplates were purchased form Costar (Corning, NY, USA). The electric heating constant-temperature incubator was purchased from Tianjin Zhonghuan Experiment Electric Stove Co. Ltd. (Tianjin, China). The automated plate washer (Wellwash 4 MK2), microplate reader (Multiskan MK3), and direct heat CO2 incubator were purchased from Thermo (Vantaa, Finland). The HPLC system consisted of a Waters 600E multisolvent delivery system and a Waters 2487 dual λ absorbance detector (Milford, MA, USA). The 0.2- and 0.45-μm syringe filters were purchased from Pall (Ann Arbor, USA).
Preparation of protein–hapten conjugates
The hapten was artesunate (Fig. 1) and was conjugated to BSA and OVA as immunogen and coating antigen, respectively. The protein conjugates were synthesized by the mixed anhydride method as previously described [22] with slight modification. Briefly, to 15 mg of artesunate in 1 mL of DMSO was added 100 μL of tri-n-butylamine. The solution was stirred for 30 min at room temperature followed by addition of 50 μL of isobutylchloroformate and stirred for another 30 min. The reaction mixture was added dropwise to 30 mg of BSA or OVA dissolved in 3 mL of 0.13 M NaHCO3; then, the solution was adjusted immediately to pH 7.5 with 1 M NaHCO3 and stirred overnight at 4 °C. The mixture was dialyzed against 2 L of 0.01-M phosphate buffer containing 0.15 M NaCl (PBS, pH 7.5) for 3 days with two changes per day, then lyophilized and stored at −20 °C.
Immunization
The protocol of immunization was similar to that described previously [26]. Five female Balb/c mice, 7 weeks old, were immunized with the immunogen. Mice were initially injected intraperitoneally with 100 μg of immunogen in 100 μL PBS emulsified with an equal volume of complete Freund’s adjuvant. Mice were subsequently injected three more times with the immunogen emulsified with incomplete Freund’s adjuvant at 2-week intervals. One week after the fourth injection, mouse antisera were collected from the retrobulbar plexus, and then the titer and specificity of each antiserum were determined by ELISAs as described below. The best-performing mouse was selected for hybridoma production and was boosted with 100 μg of immunogen in 100 μL PBS 3 days before fusion.
Monoclonal antibodies generation, purification, and characterization
The production and purification of mAbs were performed according to the procedures described previously [26]. Murine SP2/0 myeloma cells (China Institute of Veterinary Drug Control, Beijing, China) were grown in DMEM supplemented with 20% FBS, 0.2 M glutamine, 50,000 U/L penicillin, and 50 mg/L streptomycin (designated complete medium). Spleen cells harvested from the best-performing mouse were fused with SP2/0 at a ratio of 10: 1. The hybridomas were selectively cultured in complete medium supplemented with 1% (v/v) HAT for approximately 2 weeks and the supernatants were screened by ELISAs described below. Inhibition assays were performed concurrently using artemisinin, deoxyartemisinin, and the artemisinin precursors arteannuin B and artemisinic acid. The hybridomas which produced antibodies having good reactivity with artemisinin and no reactivity with deoxyartemisinin, arteannuin B, and artemisinic acid were selected. Positive hybridomas were cloned twice by limiting dilution followed by expansion for large-scale production of mAbs. The resulting mAbs were generated by inoculating selected hybridoma cells into BALB/c mice treated with mineral oil. Antiartemisinin mAbs were purified from ascite fluids by ammonium sulfate precipitation followed by protein-A-CL Sepharose 4B affinity chromatography (HiTrap, Amersham, USA). The class and subclass of the isotypes of the mAbs were determined using a mouse mAb isotyping kit. The specificity of the mAbs was evaluated by cross-reactivity with artemisinin analogs (Fig. 1) utilizing icELISA.
Indirect competitive ELISA
The protocol for icELISA was the same as that described previously [26]. Microplate wells were coated with hapten–OVA (200 μL per well in 0.05-M carbonate buffer, pH 9.6) for 3 h at 37 °C. The plate was washed four times with PBS and then blocked with 200 μL per well of 3% non-fat dry milk in PBS for 30 min at 37 °C. The plate was washed four times with PBST (PBS containing 0.1% (v/v) Tween-20); 100 μL per well of standard or analytes and 100 μL per well of antisera, supernatant, or mAbs both diluted in PBSTG (PBST containing 0.5% (w/v) gelatin, PBSTG) were pipetted and then incubated at 37 °C for 30 min. The plate was washed again four times with PBST. An aliquot of 200 μL per well of peroxidase-labeled goat antimouse IgG diluted in PBSTG was then added. After being incubated for 30 min at 37 °C, the plate was washed four times again with PBST; then, 200 μL per well of substrate solution (0.01 M citrate–phosphate buffer, pH 5.5, containing 2 mg/mL of OPD and 0.04% (v/v) H2O2) was added. The reaction was terminated by adding 100 μL of 2 M H2SO4 per well, and absorbance at 492 nm was read with the microplate reader.
Indirect non-competitive ELISA
The procedure of indirect non-competitive ELISA (incELISA) was generally the same as inELISA described above, except that the competitive step of 100 μL per well of standard or analytes and 100 μL per well of antisera, supernatant, or mAbs was replaced with 200 μL per well of antisera, supernatant, or mAbs diluted in PBSTG without any standard or analytes. Monitoring of the titer of antisera, supernatant, or mAbs and screening of positive hybridoma clones were done by incELISA.
Sample extraction
The dried samples of wild A. annua were harvested from several provinces in China. Artemisinin samples were extracted according to the method of Zhao and Zeng [28] with modifications described by Han et al. [29]. Briefly, the dried sample was powdered by a high-speed grinder and 40 mL petroleum ether (boiling range 30–60 °C) was added to 100-mg portions of the powder in a tube (Corning, NY, USA) and let sit for 16 h at room temperature. The next day it was extracted by ultrasonication (SB5200, Branson, Shanghai, China) for 2 min, followed by filtering under vacuum. The filtrate obtained was evaporated to dryness at room temperature under a mild stream of nitrogen. The residue was then dissolved in 10 mL methanol and centrifuged at 12,000×g for 5 min. The supernatant was collected as the final extract and kept at −20 °C until ELISA and HPLC analyses. The extract was diluted directly in PBSTG at a ratio of 1:5,000–10,000 for ELISA. As for HPLC, 1-mL extract was pipetted into a 10-mL volumetric flask followed by the addition of 4 mL of 0.2% (w/v) NaOH and brief mixing. After incubation at 50 °C in a water bath for 30 min, the mixture was cooled to room temperature with tap water, and then 0.05 M acetic acid was added up to a total volume of 10 mL and mixed thoroughly. The final solution was filtered through a 0.45-μm membrane prior to HPLC analysis.
Recovery test for artemisinin spiked in A. annua samples
An amount of 100-mg ground A. annua sample, of which the artemisinin content was quantified by icELISA, was spiked with artemisinin at concentrations ranging from 156 to 5,000 μg/g. The ground A. annua sample with no artemisinin added was also used as the blank control. After being kept at 4 °C overnight, the sample was extracted according to the extraction procedure for ELISA as described in the previous section. The extract was then analyzed with icELISA. Three separate extracts were taken for each spiked sample, and each extract was analyzed in triplicates.
HPLC analysis of artemisinin
Standards and A. annua samples were analyzed by HPLC according to the procedure of Zhao and Zeng [28]. The mobile phase, standards, and sample extracts obtained above were filtered through a 0.45-μm filter prior to HPLC. A C18 reverse-phase column (250 × 4.6 mm, 5-μm particle size; Thermo, Vantaa, Finland) was used to separate artemisinin. The mobile phase was 50% methanol in 0.01 M PBS (pH 7.0) at a flow rate of 1 mL/min. The UV absorption was detected at 260 nm. The injection volume was 10 μL. The retention time of artemisinin was approximately 8.1 min. All data were collected and analyzed by Waters Millennium32 software.
Results
Screening of antisera and production of mAbs
After the fourth injection, the mice were bled to collect antisera and the antisera were screened against the coating antigen synthesized by the mixed anhydride method. The titers and inhibitions of antisera by artemisinin were estimated with incELISA or icELISA. The titer was defined as serum dilution giving an absorbance of 1.0 in incELISA. The mouse with the highest titer of approximately 2.5 × 104 and the best percentage inhibition of 88% was used for further study. The hybridomas were screened with incELISA and icELISA. Three positive wells on 96-well microtiter plates were cloned twice by limiting dilution. Three clones, designated as 3H2, 3E6, and 5F9, which could secrete mAbs against artemisinin, were obtained. The mAbs 3H2, 5F9, and 3E6 were tested for their competitive binding to artemisinin in icELISA and the IC50 value of mAb 3H2 by artemisinin was remarkably lower than those of mAb 5F9 and 3E6 (data not shown). Therefore, 3H2, which could secrete mAb against artemisinin with high sensitivity and low cross-reactivity with the precursors of artemisinin, were expanded and used to produce ascites.
Characterization of mAbs
The mAb 3H2 was IgG1κ. The cross-reactivity was tested with artesunate, dihydroartemisinin, artemether, arteannuin B, artemisinic acid, and deoxyartemisinin in icELISA. The results are presented in Table 1. The cross-reactivities of artesunate, dihydroartemisinin, and artemether were approximately 650%, 57%, and 3%, respectively. No competitive inhibition was observed up to 10,000 ng/mL of arteannuin B, artemisinic acid, or deoxyartemisinin.
Competitive inhibition
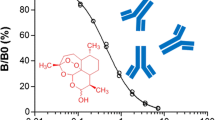
The optimal concentrations of coating antigen, mAb 3H2, and peroxidase-labeled goat antimouse IgG were screened by checkerboard titration. An icELISA under the optimized conditions was developed. Figure 2 shows a representative inhibition curve for artemisinin generated by the optimized icELISA. The IC50 value of the icELISA was approximately 1.3 ng/mL, and the working range, based on 20% to 80% of inhibition of binding of mAb 3H2 to the immobilized hapten–OVA, was 0.2–5.8 ng/mL.

Standard inhibition curve of artemisinin in icELISA format. Each value represents the mean of three replicates
Recovery of artemisinin from fortified A. annua samples by icELISA
The average recoveries for 156–5,000 μg/g of spiked artemisinin ranged from 91% to 98%, and the overall average was 95% (Table 2).
Analysis of artemisinin in A. annua samples with icELISA and HPLC
The artemisinin content in nine wild A. annua samples was determined with icELISA. The artemisinin content varied remarkably in different wild A. annua samples and ranged from 0.25 to 6.15 mg/g (Table 3). The artemisinin content of the A. annua samples from Chongqing and Guizhou Provinces in southwestern China was greatly higher than that from Shanxi and Neimenggu Provinces in northern China. The HPLC results were similar to those of icELISA (Table 3). The correlation coefficient (R 2) between the two assays was larger than 0.99.
Discussion
Analysis of artemisinin is a challenge as it possesses properties such as low UV VIS absorbance, low stability, low content, and interference of its precursors in the plant [30]. Although some conventional instrumental methods have been widely used to analyze artemisinin, they are not simple and sensitive enough to determine artemisinin in a small amount of samples obtained from plant tissues, young seedlings, cell or tissue cultures, and other biological fluids. The ELISA is highly sensitive and specific and could overcome these problems. The icELISA developed in the present study was more sensitive than HPLC-UV. Moreover, it was approximately two, six, and eight times more sensitive, based on the limit of detection, than the pAb-based immunoassays described by Jaziri et al. [22], Ferreira and Janick [23], and Song et al. [21], respectively, and approximately 10,000 times more sensitive than the mAb-based ELISA described by Tanaka et al. [24].
The artemisinin molecule lacking a reactive group is difficult to covalently link to a protein carrier directly. Ferreira and Janick [23] and Song et al. [21] derivatized artemisinin into dihydroartemisinin, dihydroartemisinin carboxymethylester, and then dihydroartemisinin carboxymethylether which the latter was finally linked to carrier protein. Jaziri et al. [22] synthesized 10-succinyldihydroartemisinin (artesunate) from artemisinin in two steps and then conjugated it to carrier protein. While Eggelte et al. [25] used artelinic acid as hapten to synthesize protein conjugates. In the present study, commercial artesunate was used as the hapten to obtain artesunate–BSA and artesunate–OVA according to the mixed anhydride method of Jaziri et al. [22] and Tanaka et al. [24] with slight modifications. In the present study, the reaction was carried out at room temperature instead of at 4 °C at which the reaction solution in DMSO solidifies (melting point of DMSO, 18.5°C). We also prepared the immunogen and coating antigen via a carbodiimide method, but the titers and inhibitions of antisera raised from the hapten–BSA synthesized by the carbodiimide method were not as good (poor binding) as those raised from the hapten–BSA synthesized by the mixed anhydride method (data not shown).
The specificity of mAb 3H2 was evaluated with artemisinin and its analogs. The cross-reactivities of mAb 3H2 with artesunate, dihydroartemisinin, and artemether were 650%, 57%, and 3%, respectively. The mAbs generated by Tanaka et al. [24] showed similar cross-reactivities, which were 630% for artesunate and 29.9% for dihydroartemisinin, while the artemisinin pAbs prepared by Song et al. [21] recognized artesunate, dihydroartemisinin, and artemether equally well. MAb 3H2 was significantly improved for specific recognition of artemisinin analogs for changes at C-10 position. Deoxyartemisinin, lacking the peroxide bridge, showed no inhibition in icELISA in the present study, which agreed with the results of Jaziri et al. [22]. The peroxide moiety was important for the antibody specificity. Eggelte et al. [25] found their mAbs raised by artelinic acid–BSA conjugate bound artemisinin and artemether (3–5% cross-reactivity) but showed low reactivity with artesunate and dihydroartemisinin, which was not explained. Since arteannuin B and artemisinic acid are two common precursors of artemisinin in A. annua [31, 32], the cross-reactivities of the antibodies against artemisinin with arteannuin B and artemisinic acid would confirm whether the ELISA was accurate enough to quantity artemisinin in the A. annua plants. Tanaka et al. [24] did not provide the cross-reactivities of their mAb with arteannuin B and artemisinic acid. MAb 3H2 had no detectable affinity with arteannuin B and artemisinic acid up to a concentration of 10,000 ng/mL. Their low affinity with mAb 3H2 attributes to the icELISA accuracy to determine artemisinin in A. annua. In addition, the high cross-reactivities with artesunate, dihydroartemisinin, and artemether might extend the usefulness of the icELISAs for pharmacokinetic studies of these compounds.
Wild A. annua samples were analyzed with the icELISA and HPLC. The dynamic range of the ELISA described by Tanaka et al. [24] was 2–20 μg/mL for artemisinin, while that of the present assay was 0.2–5.8 ng/mL. The present icELISA is far more sensitive and, thus, allows high dilution of the samples to minimize background interference. The high sensitivity can afford up to 5,000- to 10,000-fold dilutions of the samples to completely eliminate matrix interference and thus increase the assay accuracy. The results obtained from the icELISA agreed well with those from HPLC analysis with a correlation coefficient of 0.9969 (y = 1.0032x − 0.108; Fig. 3).

Correlation between artemisinin content of A. annua samples determined by icELISA and by HPLC
The results showed that the artemisinin levels differed significantly in A. annua from different provinces in China (Table 3). A. annua from Chongqing, Guizhou, and Hunan Provinces had remarkably higher artemisinin content than that from other places, which supports the fact that Chongqing, Guizhou, and Hunan Provinces are the main production areas of quality raw A. annua. Finally, it is our hope that mAb 3H2 and this simple and economical assay will be well used in malaria research and control as well as A. annua quality control, particularly in Africa and South-East Asia.
References
Li G, Guo X, Jin R, Wang Z, Jian H, Li Z (1982) J Trad Chin Med 2:125–130
Krishna S, Uhlemann AC, Haynes RK (2004) Drug Resist Updat 7:233–244
Luo XD, Shen CC (1987) Med Res Rev 7:29–52
Schmid G, Hofheinz W (1983) J Am Chem Soc 105:624–625
Xu XX, Zhu J, Huang DZ, Zhou WS (1983) Acta Chimica Sinica 41:574–576
Ro DK, Paradise EM, Quellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Keasling JD (2006) Nature 440:940–943
Pras N, Visser JF, Batterman S, Woerdenbag HJ, Malingre TM, Lugt CB (1991) Phytochem Anal 2:215–219
Acton N, Klayman DL (1985) Planta Medica 51:441–442
Zhao SS, Zheng MY (1985) Planta Medica 51:233–237
Singh A, Vishwakarma RA, Husain A (1988) Planta Medica 54:475–476
Acton N, Klayman DL, Rollman IJ (1985) Planta Medica 51:445–446
Melendez V, Peggins JO, Brewer TG, Theoharides AD (1991) J Pharm Sci 80:132–188
Ferreira JFS, Charles DJ, Wood K, Simon JE, Janick J (1994) Phytochem Anal 5:116–120
Avery BA, Venkatesh KK, Avery MA (1999) J Chromatogr B 730:71–80
Christen P, Veuthey JL (2001) Curr Med Chem 8:1827–1839
Peng CA, Ferreira JFS, Wood AJ (2006) J Chromatogr A 1133:254–258
Banthorpe DV, Brown GD (1989) Phytochemistry 28:3003–3007
Woerdenbag HJ, Pras N, Bos R, Visser JF, Hendriks H, Malingre TM (1991) Phytochem Anal 2:215–219
Woerdenbag HJ, Bos R, Salomons MC, Hendriks H, Pras N, Malingre TM (1993) Flav Frag J 8:131–137
Wang M, Park C, Wu Q, Simon JE (2005) J Agric Food Chem 53(18):7010–7013
Song ZY, Zhao KC, Liang XT, Liu CX, Yi MG (1985) Acta Pharma Sinica 20:610–614
Jaziri M, Diallo B, Vanhaelen M, Homes J, Yoshimatsu K, Shimomura K (1993) Phytochemistry 33:821–826
Ferreira JFS, Janick J (1996) Phytochemistry 41:97–104
Tanaka H, Putalun W, De-Eknamkul W, Matangkasombut O, Shoyama Y (2007) Planta Medica 73(10):1127–1132
Eggelte TA, van Agtmael MA, Vuong TD, van Boxtel CJ (1999) Am J Trop Med Hyg 61(3):449–456
Zhao J, Li G, Wang BM, Liu W, Nan TG, Zhai ZX, Li ZH, Li QX (2006) Anal Bioanal Chem 386:1735–1740
Yu FY, Lin YH, Su CC (2006) J Agric Food Chem 54:2496–2501
Zhao SS, Zeng MY (1986) Chin. J Pharma Anal 6:3–5
Han JL, Liu BY, Ye HC, Wang H, Li ZQ, Li GF (2006) J Integr Plant Biol 48:482–487
Dhingra V, Rao KV, Narasu ML (2000) Life Sci 66:279–300
Sangwan RS, Agarwal K, Luthra R, Thakur RS, Singh-sangwan N (1993) Phytochemistry 34:1301–1302
Roth RJ, Acton N (1989) J Nat Prod 52:1183–1185
Acknowledgement
We thank Prof. L. Yang, China Academy of Chinese Medical Sciences, for providing A. annua samples.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
He, SP., Tan, GY., Li, G. et al. Development of a sensitive monoclonalantibody-based enzyme-linked immunosorbent assay for the antimalaria active ingredient artemisinin in the Chinese herb Artemisia annua L.. Anal Bioanal Chem 393, 1297–1303 (2009). https://doi.org/10.1007/s00216-008-2527-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-2527-5