
Abstract
Antipsychotic drugs are frequently associated with sudden death investigations. Detection of these drugs is necessary to establish their use and possible contribution to the death. LC–MS(MS) methods are common; however accurate and precise quantification is assured by using validated methods. This study compared extraction efficiency and matrix effects using common liquid–liquid and solid-phase extraction procedures in both ante-mortem and post-mortem specimen using LC–MS–MS. Extraction efficiencies and matrix effects were determined in five different blank blood specimens of each blood type. The samples were extracted using a number of different liquid–liquid extraction methods and compared with a standard mixed-mode solid-phase extraction method. Matrix effects were determined using a post-extraction addition approach—the blank blood specimens were extracted as described above and the extracts were reconstituted in mobile phase containing a known amount of analytes. The extraction comparison of ante-mortem and post-mortem blood showed considerable differences, in particular the extraction efficiency was quite different between ante-mortem and post-mortem blood. Quantitative methods used for determination of antipsychotic drugs in post-mortem blood should establish that there are no differences in extraction efficiency and matrix effects, particularly if using ante-mortem blood as calibrator.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Antipsychotic drugs are frequently associated with sudden death investigations. Patients receiving antipsychotic drugs have been reported to be 1.4 times more likely to experience sudden unexpected death than individuals who are free from antipsychotic drugs [1–7]. Detection of these drugs is necessary to establish their use and possible contribution to the death. Reliable qualitative and quantitative detection forms the basis of a competent interpretation of the possible role of the drug in death. Increasingly, LC–MS(MS) methods are being commonly utilized for the detection of antipsychotic drugs in a wide range of tissues including blood [2–11]. LC–MS(MS) methods provide the required sensitivity and selectivity and, in contrast with gas chromatography, do not require thermal stability of the drugs in question [12]. However, accurate and precise quantification needs to be assured by the use of validated methods [13, 14]. Matrix effects can limit the usability of LC–MS(MS) methods, especially using electrospray ionization [15] Therefore, an assessment of matrix effects should be included in method validation when using LC–MS(MS) [13, 16, 17]. The most efficient approach to reduce matrix effects is elimination of the sample constituents which are responsible for matrix effects [18–20]. This can be achieved by improvement of sample pre-treatment. The most common pretreatment of specimens (i.e. blood, urine) either involves liquid–liquid extraction (LLE) or solid-phase extraction (SPE). Irrespective of whether sample pre-treatment results in 100% extraction efficiency for all analytes, improvement of the method by avoiding matrix effects is essential. Generally, SPE methods are likely to produce cleaner extracts, particularly with ante-mortem blood, plasma, or serum specimens [21–23]. However, the extraction of non-decomposed post-mortem (N-DEC) or heavily decomposed post-mortem (DEC) samples can be difficult. Another major drawback in post-mortem analysis is that validation studies have usually been performed on ante-mortem blood [24–30]. Even if matrix effects are not present and extraction recoveries high in ante-mortem blood samples, the converse can be true for post-mortem specimens (i.e. considerable matrix effects and poor extraction recoveries). Therefore, the aim of this study was to compare extraction efficiency and matrix effects using common LLE and SPE methods using different ante-mortem and post-mortem blood samples.
Materials and methods
Chemicals and reagents
Chlorpromazine, haloperidol, thioridazine, trifluperazine, and Trizma base were purchased from Sigma–Aldrich (Castle Hill, NSW, Australia). Clozapine was provided by Sandoz (Pyrmont, NSW, Australia); 9-OH-Risperidone and pimozide were obtained from Janssen–Cilag (North Ryde, NSW, Australia). Amisulpride, aripiprazole, buspirone, olanzapine, promethazine, quetiapine, risperidone, and zuclopenthixol were obtained from the Division of Analytical Laboratories, (Lidcombe, NSW, Australia). Droperidol, perphenazine, promazine, and sulpiride were provided by Australian Government Analytical Laboratories (Pymble, NSW, Australia). 1-Chlorobutane, ethyl acetate, isopropanol, methanol, and formic acid were purchased from Merck (Darmstadt, Germany) Sodium sulfate, sodium bicarbonate, and ammonium formate were provided by Ajax Finechem (Taren Point, NSW, Australia). Acetic acid was purchased from BDH Chemicals (Kilsyth, VIC, Australia). All chemicals were of analytical grade or better and water was purified using a Milli-Q Ultrapure Water System (Waters, Rydalmere, NSW, Australia).
Phosphate buffer (pH 6) was prepared by dissolving 13.72 g NaH2PO4 and 4.28 g of Na2HPO4 in 1 L water. Trizma buffer (pH 9.2) was prepared by dissolving 242 g Trizma base in 1 L water. The HPLC eluents were as follows: eluent A contained 50 mmol L−1 ammonium formate in water, adjusted to pH 3.5 with formic acid; eluent B contained 0.1% formic acid in acetonitrile.
Specimens
Drug-free ante-mortem (AM) blood samples were obtained from drug-free volunteers. Samples were collected into spray coated K2EDTA preserved plastic tubes. Post-mortem blood samples were submitted to the authors’ laboratory for routine toxicological analysis. The post-mortem blood samples were regarded as drug free if none of the existing tests showed the presence of the studied drugs in any specimen (including blood, liver, and urine). Classification of the post-mortem blood samples as N-DEC and DEC was based upon pathology description of the body from which the specimen was taken. All post-mortem blood samples were collected into plastic tubes containing 1% fluoride–oxalate. This is the standard collection tube at autopsy used across Australia and New Zealand. All blood samples were stored at −20°C prior to analysis.
Sample preparation
Liquid–liquid extraction
In a 10-mL glass tube, 0.5 mL blood was mixed with 50 μL eluent A. For comparison, either 1 mL Trizma buffer, 1 mL saturated sodium sulfate solution, or 100 mg solid NaHCO3 were added. The blood–buffer mixtures were extracted with 8 mL of three different solvents or solvent mixtures, resulting in nine different extraction procedures. The extraction solvents were: ethyl acetate, mixture of diethyl ether and ethyl acetate (50:50) or 1-chlorobutane. The samples were extracted for 30 min on a rotating wheel. After a brief centrifugation to separate layers, the solvent layer was transferred to an 8-mL disposable borosilicate tube and evaporated to dryness using a TurboVap LV Evaporation System (Millennium Science, Melbourne, VIC, Australia) at 40°C for 27 min. The residue was reconstituted in 100 μL of a mixture of eluent A and eluent B (90:10) and transferred to an autosampler vial. The final extract (10 μL) was injected into the LC–MS–MS system.
Solid-phase extraction
In a 10-mL glass tube, 0.5 mL blood was mixed with 50 μL eluent A, and 1 mL phosphate buffer. The mixture was ultrasonicated for 10 min prior to centrifugation (10 min at 1800 g). The supernatant (1 mL) was added to 4 mL phosphate buffer and loaded on to SPE cartridges previously conditioned with 3 mL purified water, 3 mL methanol, and 3 mL phosphate buffer. The loaded SPE cartridges (XtrackT, XRDAH203; UCT, Bristol. PA, USA) were treated sequentially with 6 mL purified water, 1 mL acetic acid, and 3 mL methanol. The analytes were eluted with 3 mL 84% ethyl acetate, 12% isopropanol, and 4% ammonia. The eluates were evaporated to dryness using an LV Evaporation System (Millennium Science) at 40°C for 27 min. The residue was reconstituted in 100 μL of a mixture of eluent A and eluent B (90:10) and transferred to an autosampler vial. The final extract (10 μL) was injected into the LC–MS–MS system. Solid phase extractions were automated using a Rapidtrace Solid-Phase Extraction unit 50000 (Millennium Science).
Apparatus
The LC–MS–MS system consisted of an Applied Biosystems 3200 Q-TRAP linear ion-trap quadrupole mass spectrometer (Applied Biosystems, Melbourne, VIC, Australia) with Analyst software (Version 1.4.2) and equipped with a Turbo V ion source, operated in the electron spray ionization (ESI) mode, and an Agilent Technologies (AT) 1200 Series HPLC system (Agilent, Melbourne, VIC, Australia) which consisted of a degasser, a binary pump, and an autosampler.
HPLC conditions
Gradient elution was performed on an Agilent Zorbax Eclipse XDB-C18 (4.6 mm × 150 mm, 5 μm particle size; Biolab, Clayton, VIC, Australia). The mobile phase consisted of 50 mmol L−1 aqueous ammonium formate adjusted to pH 3.5 with formic acid (eluent A) and acetonitrile containing 0.1% formic acid (eluent B). During use, the mobile phase was degassed by the integrated Agilent 1200 degasser. Before starting the analysis, the HPLC system was equilibrated for 10 min with a mixture of 90% eluent A and 10% eluent B. The HPLC system was additionally equilibrated for 4 min prior to each run. The flow rate and gradient were programmed as follows: equilibration time (−4.00 min–0.00 min) 10% eluent B, flow rate 1.4 mL min−1; 0.00–1.00 min: 10% eluent B, flow rate 1.4 mL min−1; 1.01–18.00 min: gradient increase to 100% eluent B, flow rate increase to 2.2 mL min−1; 18.01–20.00 min: 100% eluent B, flow rate 2.2 mL min−1. The column oven was set at 60°C. The autosampler was operated at room temperature; the autosampler needle was rinsed using a wash vial filled with a mixture of eluent A and eluent B (90:10).
MS–MS conditions
For detection and quantification, the following ESI inlet conditions were applied: gas 1, nitrogen (90 psi; 620.5 kPa); gas 2, nitrogen (90 psi; 620.5 kPa); ion-spray voltage, 5,500 V; ion-source temperature, 750°C; curtain gas, nitrogen (10 psi; 68.9 kPa).
The mass spectrometer was operated in the multiple reaction monitoring (MRM) mode with the following settings: collision gas was set at medium, the collision cell exit potential was 4.00 V, the dwell time was set at 15 ms. All other settings were analyte-specific and were determined using Analyst software in the quantitative optimization mode. The settings are summarized in Table 1. Q1 and Q3 were operated in unit resolution.
Extraction efficiency, matrix effects, and process efficiency
Extraction efficiencies, matrix effects, and process efficiencies were estimated in a post-extraction addition approach, as previously described [31, 32]. Three sets of samples were prepared. Samples set 1 consisted of neat standard containing the analytes at concentrations of 10 μg mL−1 in eluent B–eluent A (10:90). For preparation of samples set 2 (matrix effects), blank blood samples from 15 different sources were first extracted, as described previously, using 15 different blood samples (5× AM, 5× N-DEC, and 5× DEC). Then, the dry residues were reconstituted in 50 μL eluent A containing the analytes at a concentration of 10 μg mL−1. For preparation of samples set 3 (extraction efficiency), blank blood samples from the same sources as those in set 2 were spiked with 50 μL eluent A containing the analytes at a concentration of 10 μg mL−1. Thereafter, they were extracted as described previously, and the dry residues were reconstituted in 50 μL eluent A. Extraction efficiencies were estimated by comparison of the peak areas from the samples from set 3 with those from the corresponding samples of set 2 and reported as percentages. Matrix effects were estimated by comparison of the peak areas from the samples of set 2 with those from the corresponding samples of set 1 and reported as percentages. Hence, values below 100% indicate ion suppression whereas values above 100% indicate ion enhancement. Finally, process efficiencies (combination of extraction efficiencies and matrix effects) were estimated by comparison of the peak areas of the samples from set 3 with those from the corresponding samples of set 1.
Results and discussion
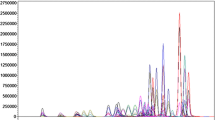
Determination of extraction efficiencies and matrix effects is a major part of LC–MS method validation according to international guidelines [13, 22, 23]. For detection of drugs in blood or plasma, sample cleanup is used to reduce the possibility of matrix effects. This study describes the comparison of commonly used LLE and SPE methods in terms of extraction efficiencies and matrix effects. Prior to mass spectrometric detection, the extracts were separated using HPLC. A representative chromatogram of the separation and detection is given in Fig. 1.

Representative chromatogram of the separation and detection of 19 antipsychotics in blood using HPLC–MS–MS
For some analytes, the concentrations selected to compare extraction efficiencies and matrix effects were not based on therapeutic concentrations. It is recognized that for postmortem blood some of these drugs will exhibit a high degree of redistribution resulting in concentrations exceeding those deemed to be therapeutic in ante mortem specimens.
Comparison of different LLE procedures
Different conditions for LLE are described in the literature. Prior to extraction of drugs with an organic solvent, blood samples are usually adjusted to a certain pH value. Additionally, use of buffer solutions with a high salt content improves the extraction efficiency as the high salt content decreases the solubility of the drugs of interest in blood. This forces the partly lipophilic drugs to be transferred into the organic phase and therefore increases the extraction efficiency. For the LLE of antipsychotics, the blood is usually buffered to a slightly basic pH, as these drugs generally have basic properties and a basic pH decreases the solubility in the aqueous blood even further. The most common buffers used for the extractions of drugs and drugs of abuse are Trizma buffer [33], saturated sodium sulfate [34, 35], or solid sodium hydrogen carbonate. Typically, the most common organic solvents for the LLE of drugs and drugs of abuse include ethyl acetate, 1-chlorobutane [36–38], or the mixture diethyl ether–ethyl acetate (50:50) [34, 35]. We therefore compared the extraction efficiencies of 19 antipsychotics and LC–MS matrix effects using these solvents and buffers.
In the first experiment, the 19 antipsychotics were extracted from spiked whole blood samples at a concentration of 10 μg mL−1 each, using nine different combinations of buffers and organic solvents. Of all the buffers used in these experiments, the highest extraction efficiencies from ante-mortem blood samples were obtained using Trizma buffer, irrespective of the solvent used. The extraction efficiencies obtained when using neutral Na2SO4 were generally lower than those obtained with basic buffer, which suggests that antipsychotics are better extracted from blood samples using basic buffer.
For the majority of drugs, 1-chlorobutane gave the highest extraction efficiencies of the three different solvents used. Sulpiride was the only antipsychotic which showed considerable lower extraction efficiency using 1-chlorobutane in comparison to the other solvents.
Considerable matrix effects in extracted blood samples were observed for olanzapine when using ethyl acetate as solvent. Otherwise, matrix effects were similar when using different buffers and solvents. However, it was observed that the use of 1-chlorobutane resulted in fewer matrix effects than other solvents.
Overall, LLE using Trizma buffer and 1-chlorobutane showed the best results in terms of extraction efficiencies and matrix effects. Therefore, this extraction method was chosen for further comparison of LLE and SPE.
Comparison of LLE and SPE
LLE extracts and SPE extracts of spiked AM, N-DEC, and DEC blood samples were compared. Five different blood samples from each group were used for this comparison.
Table 2 shows median values, standard deviations, and the range of extraction efficiencies using the LLE and SPE techniques. Data sets where the range is more than ±20% difference of the median value (not acceptable) are marked bold and underlined. With the exception of sulpiride, extraction efficiencies were comparable between LLE and SPE. For sulpiride extraction efficiencies were much lower (6% vs. 42% in AM samples) using LLE.
Comparison of AM, N-DEC, and DEC specimens shows that the median extraction efficiencies are comparable for most drugs, but the range of values determined in different DEC specimens varies. In comparison with SPE, this variation is more observable using LLE. The generally lower extraction recoveries for SPE for most analytes can be explained by the extraction procedure used. Blood from decomposed bodies can be viscous and with an oily consistency and often these samples block SPE cartridges. Despite use of extraction cartridges with a larger pore size, blockages or slow elution rates occurred using diluted blood. Therefore, all blood samples were diluted, ultra-sonicated, and centrifuged prior to loading on to the SPE cartridge. One millilitre of a total volume of 1.5 mL of diluted blood samples was used to avoid blockages. As this reduced volume used for analysis was not corrected in calculations, results for SPE can theoretically not exceed 66.6% extraction efficiency.
Table 3 shows median values and the range of matrix effects using LLE or SPE. Data sets where the range is more than ±20% difference of the median value (not acceptable) are marked bold and underlined. With some exceptions, the variations of matrix effects over five different blood samples in the same group were acceptable for AM and N-DEC samples. The application of this experiment to DEC samples showed considerable variations over five different samples, even though the median value was still comparable with the value determined in AM and N-DEC samples. The variation in matrix effects between the DEC blood samples was slightly better using SPE compared with LLE, however, these variations were still more than 20% for most analytes. Because of these considerable differences between different blood samples, methods for detection of antipsychotic drugs in post-mortem material should also consider validation experiments in blank post-mortem blood. Experiments conducted in this research did not compare preservative agents or anticoagulants pertaining to collection tubes. Further studies may be necessary to define what contribution different collection tubes make to matrix effects and extraction efficiency. To our knowledge, this is the first publication describing the differences in matrix effects of different qualities of blood samples for antipsychotics.
Conclusion
The study presented here compares extraction efficiencies and LC–MS matrix effects for 19 antipsychotic drugs in AM, N-DEC, and DEC blood. The study shows that LLE and SPE methods in ante-mortem blood are generally comparable; both extraction methods show good and reliable extraction efficiencies and low matrix effects in these samples. However, the study also shows considerable differences between clinical and post-mortem blood from decomposed bodies in terms of extraction efficiency and LC–MS matrix effects. Therefore, methods for detection of antipsychotic drugs in post-mortem material should also consider validation experiments in drug-free post-mortem blood. We also suggest that validation experiments for post-mortem analysis methods should always include extraction efficiency and matrix effect studies in N-DEC and DEC samples.
References
Jindal R, MacKenzie EM, Baker GB, Yeragani VK (2005) J Psychiatry Neurosci 30:393–395
Jones AW (2006) Toxicol Rev 25:15–35
Hoiseth G, Karinen R, Christophersen AS, Olsen L, Normann PT, Morland J (2007) Forensic Sci Int 165:41–45
Helander A, Dahl H (2005) Clin Chem 51:1728–1730
Krivankova L, Caslavska J, Malaskova H, Gebauer P, Thormann W (2005) J Chromatogr A 1081:2–8
Borucki K, Schreiner R, Dierkes J, Jachau K, Krause D, Westphal S, Wurst FM, Luley C, Schmidt-Gayk H (2005) Alcohol Clin Exp Res 29:781–787
Killian JG, Kerr K, Lawrence C, Celermajer DS (1999) Lancet 354:1841–1845
Morini L, Politi L, Zucchella A, Polettini A (2007) Clin Chim Acta 376:213–219
Kirchherr H, Kuhn-Velten WN (2006) J Chromatogr B 843:100–113
Josefsson M, Kronstrand R, Andersson J, Roman M (2003) J Chromatogr B 789:151–167
Kratzsch C, Peters FT, Kraemer T, Weber AA, Maurer HH (2003) J Mass Spectrom 38:283–295
Maurer HH (2004) Clin Chem Lab Med 42:1310–1324
Peters FT, Drummer OH, Musshoff F (2007) Forensic Sci Int 165:216–224
Maurer HH (1998) J Chromatogr B 713:3–25
Souverain S, Rudaz S, Veuthey JL (2004) J Chromatogr A 1058:61–66
Chin C, Zhang ZP, Karnes HT (2004) J Pharm Biomed Anal 35:1149–1167
Maurer HH (2007) Anal Bioanal Chem 388:1315–1325
Buhrman DL, Price PI, Rudewicz PJ (1996) J Am Soc Mass Spectrom 7:1099
Bonfiglio R, King RC, Olah TV, Merkle K (1999) Rapid Commun Mass Spectrom 13:1175–1185
Dams R, Huestis MA, Lambert WE, Murphy CM (2003) J Am Soc Mass Spectrom 14:1290–1294
Mueller C, Schaefer P, Stoertzel M, Vogt S, Weinmann W (2002) J Chromatogr B 773:47
Chambers E, Wagrowski-Diehl DM, Lu Z, Mazzeo JR (2007) J Chromatogr B 852:22–34
Matuszewski BK, Constanzer ML, Chavez-Eng CM (1998) Anal Chem 70:882–889
Robertson MD, McMullin MM (2000) J Forensic Sci 45:418–421
Roman M, Kronstrand R, Lindstedt D, Josefsson M (2008) J Anal Toxicol 32:147–155
Sporkert F, Augsburger M, Giroud C, Brossard C, Eap CB, Mangin P (2007) Forensic Sci Int 170:193–199
Gerber JE, Cawthon B (2000) Am J Forensic Med Pathol 21:249–251
Anderson DT, Fritz KL (2000) J Anal Toxicol 24:300–304
Jenkins AJ, Sarconi KM, Raaf HN (1998) J Anal Toxicol 22:605–609
Tracqui A, Kintz P, Cirimele V, Berthault F, Mangin P, Ludes B (1997) J Anal Toxicol 21:314–318
Beyer J, Peters FT, Kraemer T, Maurer HH (2007) J Mass Spectrom 42:150–160
Beyer J, Peters FT, Kraemer T, Maurer HH (2007) J Mass Spectrom 42:621–633
Rodda KE, Dean B, McIntyre IM, Drummer OH (2006) Forensic Sci Int 157:121–130
Kratzsch C, Tenberken O, Peters FT, Weber AA, Kraemer T, Maurer HH (2004) J Mass Spectrom 39:856–872
Maurer HH, Kratzsch C, Kraemer T, Peters FT, Weber AA (2002) J Chromatogr B 773:63–73
Lin SN, Chang Y, Moody DE, Foltz RL (2004) J Anal Toxicol 28:443–448
Hopenwasser J, Mozayani A, Danielson TJ, Harbin J, Narula HS, Posey DH, Shrode PW, Wilson SK, Li R, Sanchez LA (2004) J Anal Toxicol 28:264–267
Gammans RE, Kerns EH, Bullen WW (1985) J Chromatogr 345:285–297
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Saar, E., Gerostamoulos, D., Drummer, O.H. et al. Comparison of extraction efficiencies and LC–MS–MS matrix effects using LLE and SPE methods for 19 antipsychotics in human blood. Anal Bioanal Chem 393, 727–734 (2009). https://doi.org/10.1007/s00216-008-2498-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-2498-6