
Abstract
A bioanalytical detection method for specific detection of viable human pathogenic Cryptosporidium species, C. parvum, C. hominis, and C. meleagridis is described. Oocysts were isolated from water samples via immunomagnetic separation, and mRNA was extracted with oligo-dT magnetic beads, amplified using nucleic acid sequence-based amplification (NASBA), and then detected in a nucleic acid hybridization lateral flow assay. The amplified target sequence employed was hsp70 mRNA, production of which is stimulated via a brief heat shock. The described method was capable of detecting one oocyst in 10 μL using flow-cytometer-counted samples. Only viable oocysts were detected, as confirmed using 4′,6-diamidino-2-phenylindole and propidium iodide (DAPI/PI) staining. The detection system was challenged by detecting oocysts in the presence of large numbers of common waterborne microorganisms and packed pellet material filtered from environmental water samples. When the method was compared with EPA Method 1622 for C. parvum detection, highly comparable results were obtained. Since the described detection system yields unambiguous results within 4.5 h, it is an ideal method for monitoring the safety of drinking water.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cryptosporidium is a waterborne protozoan parasite, several species of which can cause cryptosporidiosis, an intestinal disease in humans and domestic mammals. In immunocompromised or immunosuppressed patients cryptosporidiosis can be fatal. Three species are known to cause infection in humans; C. parvum, C. hominis, and C. meleagridis have all been implicated as causative agents in the infection of humans, regardless of immunological status [1, 2]. Outbreaks of cryptosporidiosis have occurred on a worldwide basis from various contaminated sources, with the largest outbreak occurring in 1993 in Milwaukee, Wisconsin with over 400,000 people infected and over 100 deaths after contamination of the municipal water supply [3]. More recently, in August 2005, about 4,000 people were infected with Cryptosporidium at the Seneca Lake State Park in Geneva, New York, after chlorinated recreational water became contaminated. An estimated 300,000 cases of cryptosporidiosis occur each year in the United States resulting in approximately 66 deaths [4]. Most commonly used water treatment methods, e.g. chlorination, are ineffective at preventing transmission of Cryptosporidium as part of the parasite’s life cycle is a resilient oocyst phase. Although the number of oocysts often found in contaminated water is generally very low, a volunteer study showed that ten oocysts could cause illness in healthy people [5] and the US Food and Drug Administration (FDA) suggests that as few as one oocyst could cause infection. Therefore, a sensitive diagnostic method is necessary for effective detection.
Detection of Cryptosporidium in public water treatment systems relies on the EPA-approved Method 1622 [6]. This method requires filtration and immunomagnetic separation of oocysts from the resuspended captured material. Captured oocysts are then stained using fluorescein-isothiocyanate conjugated anti-Cryptosporidium species monoclonal antibody and a nucleic acid stain to determine oocyst numbers by fluorescence microscopy. This method, however, is unable to discriminate between the three human pathogenic species and the twelve nonpathogenic species and it cannot distinguish between viable and non-viable oocysts. Additional staining methods need to be applied in order to obtain additional information about the viability of the oocysts. Alternatively, excystation of the oocysts or infectivity assays can be performed, but they require additional expertise and are extremely time-consuming and labor intensive. Thus they are not suitable for routine water analysis [7, 8].
As only viable oocysts can cause infection, detection of only these oocysts is desirable. Messenger RNA (mRNA) is a short-lived and often inducible molecule and is optimal for distinguishing viable from non-viable organisms. In this study a mRNA for a heat-shock protein, hsp70, produced by C. parvum [9], and determined to be common to C. hominis and C. meleagridis by using the basic local alignment and search tool (BLAST), was chosen as the target sequence. As a mRNA for a heat-shock protein, this molecule will be produced in large amounts when the oocysts are subjected to elevated temperatures; we will show this mRNA is only produced in viable organisms.
Lateral flow sandwich assays using dye-encapsulating liposomes as a signal amplification system have been well established. Previously, nucleic acid hybridization-based sandwich assays using liposome technology have been reported for detection of pathogenic organisms including B. anthracis [10], Dengue virus [11], and E. coli [12]. These assays are inexpensive, rapid, specific, and simple to run.
Previously, we have reported the detection of C. parvum via its hsp70 mRNA using an electrochemiluminescence assay with the specificity and potential for the development of a much simpler C. parvum assay system. Since then, we have also reported the use of a competitive liposome-lateral flow assay that was not as sensitive as other LFAs for other nucleic acid sequences using sandwich hybridization [13]. Fritsch and colleagues have reported the detection of C. parvum via the same RNA using an electrochemical micro-vessel sensor [14].
In this work, we have integrated immunomagnetic separation directly into the assay and optimized C. parvum detection using oligo-dT mRNA isolation. We have proven the extreme sensitivity of the assay using flow-cytometer counted samples of C. parvum and have proven that the assay can detect all human-pathogenic Cryptosporidium species. Finally, we compared the detection system with EPA Method 1622.
Materials and methods
zSeveral kits were used in this study, including the Dynabeads mRNA Direct Micro Kit and the Dynabeads anti-Cryptosporidium Kit from Dynal Biotech, a division of Invitrogen (Frederick, MD, USA). Also used was the NucliSens Basic Kit amplification reagents to perform the NASBA, from bioMérieux (Durham, NC, USA). All phospholipids and cholesterol where obtained from Avanti Polar Lipids (Alabaster, AL, USA). Streptavidin and sulforhodamine B (SRB) were purchased from Invitrogen. Polyethersulfone test strip material was purchased from Pall (Port Washington, NY, USA) and nitrocellulose test strip material was obtained from Hanomy, LLC (Cheshire, CT, USA). All oligonucleotides were ordered from Operon Biotechnologies (Huntsville, AL, USA). Crypt-a-Glo FITC-labeled antibody was purchased from Waterborne (New Orleans, LA, USA).
Stock solutions of C. parvum oocysts used in this study were provided by Clancy Environmental Consultants (St Albans, VT, USA) and were from the Iowa isolate maintained at Waterborne. All samples of oocysts counted by flow cytometry were provided by the Wisconsin State Laboratory of Hygiene (Madison, WI, USA). C. hominis oocysts were provided by Dr Giovanni Widmer and C. meleadgridis oocysts were provided by Dr Sal Tzipori, both of the Tufts University School of Veterinary Medicine (North Grafton, MA, USA). C. muris oocysts were also obtained from Waterborne, via Clancy Environmental Consultants. Both samples of Giardia intestinalis and Oocystis minuta were provided by Clancy Environmental Consultants, as well as all packed pellet material from environmental water samples. Escherichia coli O157:H7 was purchased from ATCC, number 43888.
Immunomagnetic separation (IMS)
Some alterations were made to use of the Dynabeads anti-Cryptosporidium Kit, in that recommended volumes were halved. Thus, 5 mL sample volumes were used and 500 μL each of SL-A and SL-B buffers were added. Fifty microliters of anti-Cryptosporidium IMS beads were added to each tube and incubated for 90 min at room temperature under constant rotation. Each tube was then placed in a magnetic particle concentrator (MPC) for 2 min and the supernatant aspirated. The bead–oocyst complex was resuspended in 500 μL 1X SL-A buffer and transferred to a sterile nuclease-free 1.5-mL Eppendorf tube. The tubes were allowed to stand in the MPC with the magnet in place for 2 min. The supernatant was aspirated from each tube and the magnet removed. Samples being processed through the Dynabeads mRNA Direct Micro Kit were then resuspended in 100 μL nuclease-free water.
Heat shock, lysis, and mRNA isolation
One-hundred microliter oocyst samples were heat shocked at 42 °C for 20 min. The oocysts were then lysed by a freeze–thaw process [15] consisting of five cycles of freezing in an ethanol–dry ice bath and thawing in a 65 °C water bath, with each treatment lasting for 1 min.
The samples were processed using the Dynabeads mRNA Direct Micro Kit for isolation of mRNA using oligo-d(T)25 superparamagnetic beads as per the manufacture’s directions. Briefly, the samples were mixed with 100 μL lysis–binding buffer containing 100 mmol L−1 Tris-HCl pH 7.5, 500 mmol L−1 LiCl, 10 mmol L−1 EDTA pH 8.0, 1% lithium dodecyl sulfate (LiDS), and 5 mmol L−1 dithiothreitol prior to lysis and mixed with 20 μL of an oligo-d(T)25 bead solution by pipetting. The solution was gently shaken, to prevent settling, at 23 °C for 5 min before being placed on a magnetic stand and the supernatant aspirated. The beads were then washed by twice adding and aspirating 100 μL wash buffer A, containing 10 mmol L−1 Tris-HCl pH 7.5, 0.15 mol L−1 LiCl, 1 mmol L−1 EDTA, 0.1% LiDS, and twice adding and aspirating 100 μL wash buffer B, containing 10 mmol L−1 Tris-HCl pH 7.5, 0.15 mol L−1 LiCl, and 1 mmol L−1 EDTA. Finally the beads were resuspended in 5 μL nuclease-free water.
NASBA procedure
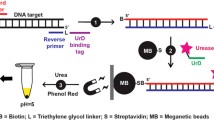
NASBA was conducted using the NucliSens Basic Kit amplification reagents as per the manufacturer’s instructions in a final volume of 20 μL. Briefly, 10 μL of the reagent mix, including 2 pmol per reaction of each of the primers shown in Table 1, were added to the 5 μL samples containing the resuspended oligo-d(T)25 beads and incubated at 65 °C for 5 min and then at 41 °C for 5 min. Five microliters of the enzyme mix was then added and the samples were returned to the 41 °C heat block for 5 min. Samples were then transferred to a 41 °C water bath for a 90-min incubation. The primers used in these experiments have previously been shown to be extremely sensitive and specific for detection of C. parvum [9].
Lateral flow assay
One microliter of NASBA product was aspirated from the sample, still containing the oligo-d(T)25 beads, and mixed with 2 μL liposomes tagged with the reporter probe shown in Table 2, prepared as previously reported [16], and 5 μL hybridization buffer containing 20% formamide, 4X SSC, 0.2% Ficoll type 400, and 0.125 mol L−1 sucrose, and allowed to incubate at 41 °C for 20 min. A lateral flow test strip with the capture probe (Table 2) immobilized was inserted into the tube and after the mixture migrated into the test strip an additional 35 μL of the running buffer was added to complete the assay. Signals were then read at the capture zone and a background reading was taken just below this zone using an ESECO handheld Biosmart reflectometer BR-10, λ = 560 nm (Cushing, OK, USA). Capture and background readings were subtracted to yield the final signals. The probes used in this assay have previously been shown to provide sensitive and specific detection of C. parvum [9]. Test strips were prepared as previously described using a Camag Linomat IV for consistent application of the streptavidin-capture probe solution [11, 17], with 60 pmol biotinylated capture probe and 20 pmol streptavidin immobilized on each test strip. A positive signal is defined as a reflectometer reading at the capture zone that is more than 5 above the background reading, with any sample yielding a difference less than this considered to be negative.
Verification of viability detection by 4′,6-diamidino-2-phenylindole and propidium iodide (DAPI/PI) staining
A stock sample containing 5.9 × 104 oocysts of C. parvum per 100 μL was split to provide four aliquots of 1 mL each in screw-top, gasket-sealed microcentrifuge tubes. Of these aliquots, two were placed in boiling water for a total of 15 min, removing every 5 min to vortex mix. All four samples were then allowed to rest at room temperature for 72 h to allow any hsp70 mRNA produced at the early stages of boiling to degrade.
One boiled sample and one control sample were centrifuged at 12,000 rpm for 1 min in a microcentrifuge and the supernatant was aspirated, leaving the pellet and less than 50 μL supernatant. One milliliter of acidified Hanks’s balanced salt solution (HBSS), pH 2.75, was added to the samples and they were allowed to incubate at 37 °C for 1 h. The samples were centrifuged as before, the supernatant removed, and 1 mL normal HBSS added to resuspend the pellet. This wash step was then repeated and the sample was centrifuged and the supernatant aspirated once again, and the pellet was resuspended in 100 μL normal HBSS.
To each sample, 10 μL 2 mg mL−1 4′,6-diamidino-2-phenylindole (DAPI) in methanol and 10 μL 1 mg mL−1 propidium iodide (PI) in 1 × PBS, pH 7.2 were added. The samples were then incubated in the dark for 90 min at 37 °C. Seven microliters of a 1× working solution of Crypt-a-Glo FITC-labeled antibody was added to each sample and it was allowed to incubate for another 30 min at 37 °C. Upon removal from incubation 1 mL normal HBSS was added to each sample and vortex mixed.
Slides were prepared by placing 5 μL of a sample on each slide and covering with a 18-mm2 coverslip, which was then sealed with clear nail-polish. Samples were then examined at 600× using the appropriate filters and DIC optics in order to classify oocysts based on inclusion or exclusion of DAPI and PI, and visualization of the contents of the oocysts as shown in Table 3.
Boiled samples were to be examined until a minimum of 200 oocysts were counted and non-boiled control samples were examined to confirm the presence of viable oocysts.
These samples were then compared with the corresponding boiled and non-boiled samples, which where heat shocked and lysed as described. The samples were then diluted from the original number of oocysts to ten oocysts per 100 μL and divided into 100 μL aliquots prior to mRNA isolation, amplification, and detection as described.
Preparation of environmental water samples
Environmental water samples were filtered as per method 1622 and all material captured on the filter was eluted and collected by Clancy Environmental Consultants. Each sample was characterized based on biological and inorganic contaminants, as shown in Table 4. Some of the samples were shipped containing formalin. In order to wash out any formalin, each of the environmental water samples was centrifuged for 20 min at 1,200 g, the supernatants were removed, and the packed pellet volume estimated. Each sample was then diluted to 5% packed pellet volume prior to being split into replicates. Environmental water samples were spiked into tubes containing 100 oocysts of C. parvum in 500 μL nuclease-free water, sorted by flow cytometry, and the total volume was brought to 5 mL with nuclease-free water, providing packed pellet volumes of 1 to 4.5% per replicate.
Results and discussion
Detection of human pathogenic Cryptosporidium spp.
Samples containing 0, 5, 25, 50, 100, 250, and 500 oocysts of C. parvum, C. hominis, or C. meleagridis in 100 μL nuclease-free water were diluted from a stock sample containing 5 × 103 oocysts mL−1 and heat shocked, lysed, and the mRNA isolated as described above. The samples were then amplified via NASBA and quantified using the lateral flow assay. All three species were successfully identified at numbers as low as five oocysts (Table 5). Since samples were generated via serial dilution from a stock solution containing 5 × 103 oocysts mL−1 and not counted via flow cytometry, the numbers of oocysts shown here are not as accurate as those measured later. However, the data demonstrate that all three human-pathogenic species of Cryptosporidium can be detected with the biosensor assay using the same primers and detection probes.
Initial experiments employed total RNA isolation using the Qiagen RNeasy Mini Kit. However, as the target molecule is mRNA and Cryptosporidium is eukaryotic, mRNA isolation using oligo-d(T)25 magnetic beads was also investigated and found to be better, because of the more specific isolation of mRNA molecules via their poly-A tail and elimination of potentially contaminating rRNAs and tRNAs. Thus, this procedure yielded more consistent results, especially for small numbers of oocysts, and was employed in all the experiments described here.
Specificity for human pathogenic Cryptosporidium
Eight samples containing 500 oocysts of C. muris in 100 μL nuclease-free water were treated as described above and amplified with a negative control and a positive control, a sample containing mRNA isolated from 500 oocysts of C. meleagridis in 100 μL. The negative control and all eight samples containing oocysts of C. muris produced negative signals on the lateral flow test strips while the positive control generated a strong positive signal at the capture zone. This indicated that no false positive signals will be generated by non-human pathogenic C. muris.
Detection in the presence of contaminating organisms
Samples containing ten oocysts, diluted from a stock of 5 × 103 oocysts mL−1, of C. parvum and approximately 5 × 104 cells of E. coli O157:H7, Giardia intestinalis, or Oocystis minuta in 5 mL were processed using the IMS procedure described above. The samples were then heat shocked, lysed, isolated, and amplified as described above. Each combination of contaminating organism and C. parvum was analyzed in quadruplicate and compared with samples containing ten oocysts of C. parvum aliquotted from the same stock run in parallel.
The samples containing C. parvum oocysts and contaminating organisms tested positive in all cases, as shown in Fig. 1. One sample out of four containing only ten oocysts of C. parvum yielded a negative result. It is assumed that either this sample was not handled correctly, or that fewer than five viable oocysts were present, because samples were generated via dilution only. Samples containing only the contaminating organisms produced no signals, confirming BLAST results which showed no potential for cross-reactivity of the primers and probes employed. Additionally, samples containing E. coli, Giardia, or a variety of other organisms were tested previously in mixed cultures with C. parvum, yielding similar results, and in pure cultures showing no false positives [9].

Cryptosporidium detection in the presence of contaminating organisms. Each sample was analyzed in quadruplicate and contained ten oocysts of C. parvum. Those containing G. intestinalis or O. minuta had 54,000 cells and samples with E. coli O157:H7 had 8.6 × CFU
Analytical sensitivity
Samples containing 1, 2, 3, 4, 5, or 10 oocysts of C. parvum in 10 μL, counted into tubes by flow cytometry, were obtained from the Wisconsin State Laboratory of Hygiene. The oocysts were taken from a sample that showed 98.7% viability prior to sorting. Prior to shipping, these samples were heat shocked as described and 100 μL of the lysis/binding buffer from the Dynabeads mRNA Direct Micro Kit were added prior to lysis. This buffer contains 5 mmol L−1 dithiothreitol, an RNase inhibitor, which should increase the stability of the target hsp70 mRNA. Samples were then shipped on dry ice, and on arrival all samples were thawed. RNA was isolated, and amplified as previously described. As shown in Fig. 2, all eight of the samples containing one oocyst of C. parvum scored positive. Additionally, all eight replicates containing 2, 3, 4, 5, and 10 oocysts scored positive (membranes look like those shown in Fig. 2 and are, therefore, not also shown here); indicating that our assay is capable of detecting a single oocyst. Thus, assuming a single oocyst present in a 5 mL sample is captured by IMS, the described method would be capable of detecting it as the final volume following IMS is always the same.

Lateral flow test strips from one oocyst C. parvum samples. Samples containing one viable oocyst of C. parvum were prepared by flow cytometry and processed through heat shock, lysis, mRNA isolation, and NASBA amplification, yielding strong signals
Discrimination between viable and non-viable oocysts
On examining the boiled sample, 315 oocysts were counted, all of which scored DAPI−/PI+ with no oocysts scoring in any other category, yielding a 0% viable score. Examination of the control sample yielded 50 oocysts scoring DAPI+/PI−, with the DAPI staining localized to the nuclei indicating that there are in fact viable oocysts in the control. As staining was only to confirm the presence of viable oocysts, counting was limited to this category even though DAPI−/PI− oocysts could be viable. It should be noted that during counting no PI+ oocysts were noted, though several DAPI− oocysts were observed. Examples of the stained oocysts from both samples are shown in Fig. 3.

DAPI/PI viability staining results. The images from the boiled sample show a cluster of six oocysts stained with (a) FITC-labeled antibody on the oocyst wall and (b) PI inclusion in the cytoplasm. It is evident from the FITC-labeled antibody (a) that there is deformation of the oocysts common in dead oocysts, note the brighter locations indicating crumpling of the oocyst walls. The control sample yielded viable oocysts; shown (c, d) are two different oocysts that showed DAPI staining localized to the nuclei. A DAPI− oocysts can be seen in (d) next to a DAPI+ oocyst
From both the boiled and the control non-boiled samples, 24 aliquots of 10 oocysts per 100 μL were processed through mRNA isolation and NASBA along with three positive and three negative controls. All 24 aliquots from the boiled sample provided no signal, correlating with the 0% viability assessed via DAPI/PI staining. Also, all 24 aliquots of the control non-boiled sample yielded positive signals, which also correlates with the observed viable oocysts via DAPI/PI staining.
Comparison with EPA Method 1622
Samples containing nominally 10 oocysts or 25 oocysts of C. parvum in 10 mL were prepared by dilution from a stock of 5 × 103 oocysts mL−1 and half were processed through the IMS, staining, and enumeration procedures of EPA Method 1622 by Clancy Environmental Consultants [6]. The remaining samples were heat shocked, had 100 μL lysis/binding buffer added, and where lysed via the previously described freeze–thaw procedure. These samples were then shipped frozen, thawed, and mRNA isolated and amplified via NASBA prior to being quantified using the lateral flow assay. The actual numbers in the samples was determined by staining and counting additional samples from the same IMS procedure, as would normally be done at the end of Method 1622 [6]. The actual number was 4 to 11 oocysts for those labeled to contain 10, and 17 to 32 oocysts for those labeled to contain 25.
The results shown in Table 6 indicate that the detection system described here is highly comparable with EPA Method 1622. While it cannot provide quantitative results, i.e. only a yes/no answer, it is very rapid, easy to perform, and does not require expensive equipment other than a freezer and a heating bath. The signal values obtained with the biosensor were lower than expected when compared with all previous analyses. We assume either that the oocysts contained in the sample were not all viable (thus reducing the number of detectable oocysts for the biosensor close to or below the detection limit) or that the two-step detection process in which samples were shipped frozen prior to RNA extraction and amplification resulted in partial loss of RNA from the samples.
Spiked environmental water samples
All four replicates of the control sample, consisting of 100 counted oocysts in 5 mL nuclease-free water, yielded positive signals as did all four replicates of samples 201–1, 243–2, 270–3, 256–6, and 094–3. This result, shown in Fig. 4—all samples tested yielding positive results—indicates that the system described can be used to detect viable C. parvum oocysts in concentrated pellet material from environmental water samples following IMS.

Lateral flow test strips for environmental water samples. Samples containing 100 oocysts counted by flow cytometry were spiked with packed pellet material in a total volume of 5 mL and processed through IMS. All samples yielded positive signals
Conclusions
Employing IMS, heat shock, freeze–thaw lysis, mRNA isolation, NASBA amplification and a nucleic acid hybridization lateral flow sandwich assay, the described method specifically detects as few as one oocyst of a viable human pathogenic Cryptosporidium species. Assuming a single oocyst present in a 5 mL sample is captured by IMS, the described method would hence be capable of detecting one oocyst per 5 mL as proven here. In addition, because large environmental water samples were filtered and used as sample matrix, we can safely assume that one oocyst captured through filtration and IMS from large samples (hundreds to thousands of liters of water) can indeed be detected with this method. Detection of these oocysts is possible in the presence of large numbers of microorganisms commonly found in contaminated water samples and in packed pellet material collected from environmental water samples. The entire method, from IMS to readable signals on the test strips, takes only 4.5 h with IMS running 90 min, a 20-min heat shock, 10 min for freeze–thaw lysis, 5 min for mRNA isolation, 115 min for NASBA (including steps prior to incubation), a 20-min liposome-target hybridization incubation, and 10 min for the lateral flow assays to produce signals. Most importantly, results obtained with the method compared very well with those from EPA Method 1622.
References
Yagita K, Izumiyama S, Tachibana H, Masuda G, Iseki M, Furuya K et al (2001) Parasitol Res 87:950–955
Leoni F, Amar C, Nichols G, Pedraza-Diaz S, McLauchlin J (2006) J Med Microbiol 55:703–707
Wilkinson SL (1997) Chem Eng News 75:24–33
Mead P, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C et al (1999) Emerg Infect Dis 5:607–625
Chappell CL, Okhuysen PC, Sterling CR, DuPont HL (1996) J Infect Dis 173:232–236
Environmental Protection Agency. Method 1622: Cryptosporidium in water by filtration/IMS/FA. Vol., 2005:45–60
Robertson LJ, Campbell AT, Smith HV (1993) Parasitology 106:13–19
Hou L, Li X, Dunbar L, Moeller R, Palermo B, Atwill ER (2004) Appl Environ Microbiol 70:642–646
Baeumner AJ, Humiston M, Montagna RA, Durst RA (2001) Anal Chem 73:1176–1180
Hartley HA, Baeumner AJ (2003) Anal Bioanal Chem 376:319–327
Baeumner AJ, Schlesinger NA, Slutzki NS, Romano J, Lee EM, Montagna RA (2002) Anal Chem 74:1442–1448
Baeumner AJ, Cohen RN, Miksic V, Min JH (2003) Biosens Bioelectron 8:405–419
Esch MB, Baeumner AJ, Durst RA (2001) Anal Chem 73:3162–3167
Aguilar ZP, Fritsch I (2003) Anal Chem 75:3890–3897
Stinear T, Matusan A, Hines K, Sandery M (1996) Appl Environ Microbiol 62:3385–3390
Edwards KA, Baeumner AJ (2006) Anal Bioanal Chem 386:1335–1343
Baeumner AJ, Pretz J, Fang S (2004) Anal Chem 76:888–894
Acknowledgement
This study was funded in part by EPA Contract Number EP-D-06–034, NYSTAR. This research was also supported in part by the Cornell University Agricultural Experiment Station federal formula funds, Project No. 123–314 received from Cooperative State Research, Education and Extension Service, US Department of Agriculture. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the US Department of Agriculture. The authors would like to thank Jennifer Clancy and Randi McQuin of Clancy Environmental Consultants, Inc., Becky Hoffman and Martin Collins of the Wisconsin State Laboratory of Hygiene, and Giovanni Widmer and Sal Tzipori of Tufts University School of Veterinary Medicine for providing samples used in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Connelly, J.T., Nugen, S.R., Borejsza-Wysocki, W. et al. Human pathogenic Cryptosporidium species bioanalytical detection method with single oocyst detection capability. Anal Bioanal Chem 391, 487–495 (2008). https://doi.org/10.1007/s00216-008-1967-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-1967-2