
Abstract
A microstructural, mineralogical, and chemical study of the nacre–prisms boundary in the shells of Pinctada margaritifera shows that this boundary is not an abrupt transition, but that there exists a distinct fibrous layer with clear topographic structures and evidence of growth lines. A three-step biomineralization process is proposed that involves changes in the chemical and biochemical composition of the last growth increments of the calcite prisms, formation of the fibrous layer, and development of regular tablets in the nacreous layer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mollusc shells are commonly described as a two-layered structure: an outer layer composed of calcite prisms and an internal nacreous layer. This long-standing oversimplification is due to the remarkable appearance of the few species in which this simple structural arrangement is illustrated. The Pelecypods, belonging to the genus Pinctada, the “pearl oysters”, are the best known. Among them, Pinctada margaritifera produces a large shell (up to 30 cm wide). The calcite prisms of the external layer are easily visible as units perpendicular to the shell surface. This organization of the external layer is in clear contrast to the finely layered nacreous aragonite with tablets parallel to the shell surface. Additionally, the pigments may contribute to a simplified view of the shell organization: usually calcite prisms are black, whereas the nacreous layers are white with highly reflective structures. Thus, both the structural and the chromatic elements within the shell suggest the existence of a sharp boundary between what are basically two different shell layers.
In fact, such a simple nacre–prisms arrangement is actually rare among the mollusc shells. Sometimes, more than two layers are present, as in the gastropod Haliotis tuberculata, which is a typical example of a three-layered shell [1–3].
From a biological standpoint, such a simple view of the bilayered shell raises questions about the mechanism that governs the biomineralization process, and how two so different materials, from mineralogy to microstructure, can be so closely joined together.
Early observations by Wada [4] had suggested that a transitional material may exist between the prismatic and nacreous layers of Pinctada martensi. This material has a spherulitic appearance, the thickness of each spherule layer being about 5–10 μm (see Figs. 10, 11 in [4]). According to Wada, “These spherules seem to be calcite grains...”. Recent techniques in submicron imaging and analysis now enable us to obtain detailed structural observations and in situ biochemical characterizations of this crucial step in nacreous layer crystallization.
Additionally, with developments in molecular biology, and specifically the construction of complementary DNA libraries [5] we can characterize directly the gene activity within cells of the ectodermal layer of the mantle. Recent results have demonstrated the high specificity of proteins involved in the mineralization process as belonging to the calcite-producing or aragonite-producing domains, and the biochemical complexity of the mineralizing process. Consequently, the macroscopically visible “sharp boundary” between the calcite and aragonite domains needs to be examined in more detail in order to identify and characterize more precisely the biochemical activity of the mantle cell layer and the resulting mineralogical transformations that occur outside the cell layer.
Therefore, the aim of this study was to improve our knowledge of the precise distribution of chemical elements, and changes in distribution of the organic components of the mineralizing matrices, at the interface between the calcite prismatic layer and the newly formed layers of the nacreous aragonite in the black-lip pearl oyster Pinctada margaritifera.
Material and methods
Material
Specimens of Pinctada margaritifera (Linnaeus, 1758) (Pteriomorpha, Pterioida, Pteriacea, Pteriidae) were collected in French Polynesia. The shell is large (25 cm), both valves are of almost equal size, subcircular in outline (Fig. 1a), and greyish-green in colour. The inner part of the shell is white with black margins (Fig. 1a).

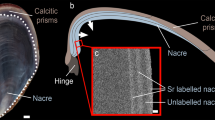
Inner view of a valve of the black-lip pearl oyster Pinctada margaritifera. a The main part of the shell is composed of the nacreous layer, and the outer edge of the outer calcite prismatic layer. b Detail of the boundary between the two layers. c Scanning electron microscope (SEM) image showing the large calcite prisms and the ridges of the nacreous layer. d View of an open shell showing the retracted mantle, not attached to the pallial line. e Vertical section showing the persistence of the boundary layer between prisms and nacre. f Detail of the fibrous layer. g Growth lines in the fibrous layer. N nacreous layer, P prismatic layer, M mantle, FA fibrous aragonite, single-headed arrows limit of the two layers, double-headed arrow growth lines
Samples were embedded in resin, polished using various grades of diamond paste down to a final 0.25 μm grade. Samples were cleaned with a detergent mixed with hot water for 1 min to remove any oil residue, and rinsed with tap water. Depending on the technique subsequently employed, additional preparative procedures were undertaken, the details of which are given later in the descriptions of the related methods.
Scanning electron microscope
Fractured and polished sections were prepared, cut perpendicular and parallel to the surface of the shells. Polished sections and some fractures were etched with various acids and enzymes (such as pronase) to reveal microstructural features. The detailed procedures of the sample preparations are given in the figure legends. Scanning electron microscope (SEM) observations were conducted using a Philips 505 instrument and a Philips XL30 instrument housed at the Laboratoire de Géologie, Université d’Orsay Paris XI.
Atomic force microscope
The atomic force microscope (AFM) maps the surface topography of the sample, where very small (nanoscale) variations in height are represented as digital images. In phase imaging, a variant of tapping mode, the phase lag of the cantilever oscillation relative to the signal sent to the cantilever’s piezo driver is used as a basis for image generation. Phase images can be generated as a consequence of variations in material properties, including composition, hardness, friction, and adhesion. AFM observations were conducted at room temperature and in air using a Digital Instruments (Veeco) Nanoscope III Dimension 3100 instrument housed at the Laboratoire de Géologie, Université d’Orsay. The probe consists of a cantilever with an integrated silicon nitride tip (Digital Instruments). Micron-scale images were acquired using the tapping mode. The detailed procedures of the sample preparations are given in the figure legends.
Electron microprobe analyses
A Cameca SX50 wavelength-dispersive electron microprobe at the Natural History Museum (NHM), London, was used to obtain the compositions and elemental maps of the samples. For quantitative analysis, the operating conditions were 15-kV accelerating voltage, 20-nA specimen current with a defocused beam, and for mapping a 100-nA specimen current was employed. Additional data were obtained using a JEOL LV5900 analytical variable-pressure SEM with an Oxford INCA energy-dispersive spectrometer at the NHM (London).
Confocal microscope
Studies were undertaken using a Leica TCS-NT SP1 confocal microscope at the NHM, London. Initial tests were carried out using the microscope’s wavelength scan function which produces a signal intensity plot for each excitation wavelength over a range of detected wavelengths. These plots were used to optimize the choice of the excitation laser, and to determine the detector window settings to use for each channel. The plots also assisted in determining the optimum settings for autofluorescence and for reflection mode, and the two laser excitation wavelengths and detector windows were set to minimize interferences.
A 488 nm argon laser was chosen for excitation with a detector window set for fluorescein isothiocyanate type emission (515–545 nm). A 633 nm helium–neon laser was used with a detector window set at 625–641 nm to detect reflection from the crystalline shell structure at the same spatial position. The choice of objective lens was dependent on the field of view and depth (z-step) resolution required; where oil immersion lenses were required, cover slips were mounted onto the sample using immersion oil. After each observation, the cover slips were removed and the samples were wiped clean of immersion oil.
Fourier transform IR microscopy
The synchrotron IR microscopy was performed on the IR end-station of the ID21 beam line at the European Synchrotron Radiation Facility (ESRF). This beam line is equipped with a Continuum IR microscope (Thermo Nicolet) coupled to a Nexus Fourier transform IR bench (Thermo Nicolet). The microscope operates in confocal mode, where the focusing Schwarzschild objective and the collection Schwarzschild objective have a magnification of ×32 (numerical aperture 0.65). Owing to its reduced source size and high collimation properties, a synchrotron IR source is more efficiently coupled to the low acceptance of the confocal microscope, while its higher spectral luminescence (brightness) allows signal-to-noise ratios to be kept at diffraction-limited resolutions. The microscope is equipped with a computer-controlled x/y stage allowing acquisition of profiles or maps of the sample. Maps were scanned with steps of 8 μm in both directions, with an aperture size of 8 × 8 μm2. Spectra were collected in reflection mode, with a resolution of 8 cm−1. For each spectrum, 256 scans were accumulated in the wavenumber range from 4,000 to 700 cm−1. The final format of the data was recorded as absorbance values.
NanoSIMS
Samples were cut perpendicular to the interface between the nacreous and the prismatic layer, mounted in epoxy (Körapox©), and polished to a 0.25-mm finish using diamond paste. The samples were then coated with gold. Chemical mapping was carried out with the Cameca NanoSIMS N50 at the Muséum National d’Histoire Naturelle in Paris. With use of a focused primary beam of Cs+, secondary ions of 12C−, 12C14N−, and 32S− were sputtered from the sample surface and detected simultaneously (in multicollection mode) using electron-multipliers at a mass-resolving power of approximately 4,000 (M/DM), sufficient to resolve all isobaric interferences. During Cs+ bombardment, charge compensation was achieved by the simultaneous delivery of electrons to the surface. Chemical maps were obtained by rastering the primary beam across a selected and presputtered sample surface. The spatial resolution of the primary beam was about 75 nm. Two zones, sited very close to each other, were mapped.
Micro X-ray absorption near edge structure spectroscopy
This work was carried out at the ID21 beamline of the ESRF. The ID21 scanning X-ray microscope uses Fresnel zone plates as focusing optics to generate a submicron X-ray probe, which is used to investigate the sample with various contrast mechanisms (fluorescence, transmission, phase contrast). An energy-dispersive high-purity Ge detector (Princeton, Gamma-Tech) mounted in the horizontal plane perpendicular to the beam collects the fluorescence photons. This geometry minimizes the contribution of elastic scattering. Provided the sample is thin enough, a Si photodiode can also be mounted downstream from the sample to exploit the transmission signal. An energy range between 2 and 7 keV is available, which gives access to the K-edge of sulphur at 2,472 eV. The X-ray absorption near-edge structure (XANES) energy scan around the sulphur K-edge is achieved using a fixed-exit double-crystal Si(111) monochromator located upstream from the microscope, which offers the necessary energy resolution. This experiment required the X-ray microscope to be operated under a vacuum to avoid the strong absorption of the sulphur emission lines by air.
Although the primary beam energy was set around that of the sulphur K-edge energy region, elements with absorption edges at lower energies were also subject to excitation and emission of fluorescence photons, and could therefore be determined. Thus, microfluorescence element maps of Mg and Na and Sr were obtained simultaneously with the S maps.
Samples were cut perpendicular to the shell surfaces. They were embedded in resin, and polished using various grades of diamond paste. The oil residue from the diamond paste was removed with a detergent diluted in hot water for 1 min, then rinsed with tap water. Samples were then etched with 1% v/v acetic acid for 5 s. Because fluorescence was used as a contrast mechanism, it was essential that the sample offered a flat surface to the X-ray beam, in order to avoid signal fluctuations that could have been produced by sample topography. Additionally, etching of the sample after polishing helped to eliminate potential surface contamination and remains of the diamond pastes and oil.
Results
Microstructures and nanostructures
The topographical relationship and extension of the two main layers at the growing edge are shown in Fig. 1: the main part of the internal surface of the valve is composed of the nacreous layer, with the calcite prismatic layer visible only along the outer edge of the shell (Fig. 1a–d). As shown by an open shell (Fig. 1d), the mantle is not attached to the pallial line, in accordance with the general characteristics of Pteriomorpha and Pteriacea [6], such that under stress the mantle is retracted and a part of the nacreous layer is visible. The boundary between the prisms and nacre appears to be thin and straight at low magnifications, and persists along the length of the shell (Fig. 1e). It should be noted that this boundary is different from the myostracum layer. Higher magnifications of vertical sections show the fibrous structure and growth lines in this layer between nacre and prisms (Fig. 1f,g).
During life, the bivalve shell increases in both length and thickness; thus, there is a progressive overlap of the calcite layer by the nacreous aragonite layer (Fig. 1a,b, arrows). However, the first elements are not deposited directly on the calcite surface of the prisms. Before aragonite is deposited, an organic layer covers previous calcite growth layers, resulting in closure of the calcite compartment of the shell (Fig. 2a). Vertical fractures through the two main shell layers clearly show the continuity between the prism envelopes and the organic layer covering the top of the calcite prisms (Figs. 2d–f, 6b,c). Figure 2b illustrates how precisely the first patches of aragonite are deposited exclusively onto the organic layer, avoiding those parts of the calcite prisms that have not yet been covered by the spreading organic layer.

Nacre–calcite prisms transition: all secondary electron SEM images. a Unetched inner surface of a valve showing the first units (fibrous aragonite) on a thick organic layer and the underlying prismatic layer (P). b Detail of the same showing the organic layer. c Detail showing the composite structure of the first units. d Etched vertical section showing the prisms (P), the nacre (N), and the fibrous aragonite (FA). Pronase 1 mg ml−1 N-(2-hydroxyethyl)piperazine-N′-ethanesulphonic acid buffer for 4 h at 38 °C, pH 7.4. e Polished and etched section showing the thick organic layer between the prisms (P) and the first layers (FA). Formic acid (5%) for 10 s. f Fixed and etched structure of the fibrous aragonite and the thick organic wall. Glutaradehyde (12%) + acetic acid (1%) + alcian blue at pH 2.5 for 5 min at 20 °C. g Detail of the fibrous structure. h Fracture showing that elongated units of the fibrous aragonite are composite. Sodium hypochlorite 12 °C for 5 min. i Alternation of “nacreous” layer and fibres in the transition. Same sample images f and g
Remarkably, these first aragonite components do not exhibit the typical morphology of the nacreous tablets. At high magnifications, SEM images show the granular appearance of this aragonite (Fig. 2c), which progressively develops into fibres organized in a fanlike arrangement (Fig. 2d–g). Between the two main shell layers this early stage of aragonite deposition creates an intermediate zone, with a growth zonation parallel to those of the nacreous and prismatic layers, but with a structure that is neither prismatic nor nacreous. This is termed here “fibrous aragonite”. The thickness of fibrous aragonite is variable (Fig. 2e–i).
At the nanometre scale, AFM imaging shows that fibrous aragonite is composed of rounded irregular-shaped nanograins only a few tens of nanometres in diameter (Fig. 3), surrounded by a black layer. Phase and amplitude images suggest that these layers are organic envelopes, and that the granules are composite features (Fig. 3e,f). The size, shape, and arrangement of the granules appear to be more irregular than those of the nacreous layer, but no statistical data are yet available.

Nacre–calcite prisms transition: atomic force microscope images. a Polished vertical section showing the granular structure of the fibrous aragonite. Milli-Q water for 20 h at 20 °C. Phase image. b Another pattern in the same sample. Phase image. c Closeup of the transition. In height image, the limits of the crystallites are not so visible. Milli-Q water for 20 h at 20 °C. d Phase image of the same. e Detail of the same showing organic envelopes (stars) surrounding the granules. Milli-Q water for 20 h at 20 °C. Phase image. f Amplitude image of the same
Elemental composition
The average concentrations of some minor elements in the nacreous and prismatic layers are shown in Fig. 4a. The Mg and S concentrations of the calcite prismatic layer are significantly higher than those of the nacreous layer, whereas Sr concentrations are similar in both the prismatic and the nacreous layers, but are lower than that of a nonbiogenic aragonite (sedimentary aragonite typically contains more than 9,000 ppm Sr; sponges and corals contain 7,000–9,000 ppm Sr; whereas mollusc shells typically contain only 1,000–2,000 ppm Sr) [7, 8]. A profile for several elements across the nacre–prisms transition is shown in Fig. 4b. The high Mg concentrations of the calcite prismatic layer abruptly decreases toward the nacreous layer, whereas the Na profile is irregular. Sr concentrations are similar in the two layers, and S concentration slowly decreases from the calcite towards the aragonite. Element distribution maps illustrate the differences in composition between the nacreous and prismatic layers, and the fibrous layer (Fig. 5). The C map shows clearly the interprismatic organic walls (Fig. 5b), and the organic layer between the calcite prisms and the fibrous layer. In all the maps (Fig. 5c–f), the composition of the fibrous layer is similar to that of the nacreous layer, and is different from that of the calcite prisms. A distribution map for S confirms the high content of S in the calcite layer (Fig. 5f). The maps also show that the prisms are not homogeneous when seen in longitudinal section, and in the region closest to the nacreous layer, there is some variation in Sr, P, and S (Fig. 5d–f).

Chemical composition. a Average contents of minor elements of the nacreous (N) and prismatic (P) layers. b Profile from the prismatic to the nacreous layer, showing changes in Mg, Sr, and S concentrations

Compositional maps. a Backscattered electron image showing the region analysed. b C map showing the organic composition of the interprismatic walls. c Mg map. d Sr map. e P map. f S map. All the maps show the similarity of composition between the fibrous layer and the nacre. N nacre, P prisms, FA fibrous layer, w organic interprismatic wall
From the element distribution maps in Fig. 5, it can also be seen that the fibrous layer is similar to the aragonite nacreous layer.
Organo-mineral relationships
Confocal microscope images of the growing edge of the inner surface of the shell show the compositional differences between the thick organic walls surrounding the prisms and the inner part of the prisms (Fig. 6a). Prisms appear as homogeneous structures, with no visible intraprismatic organic membranes. Then, it can be seen that the fibrous (fibrous aragonite) granules are first secreted on an organic layer similar to that of the interprismatic walls. Vertical sections also show the thick organic interprismatic walls, with their typical undulating outlines (Fig. 6b). These undulations reflect the growth lines present within the interprismatic organic walls. Growth lines are visible in both fluorescence and reflection images. They cross the adjacent prisms, showing the cyclical secretion process has occurred throughout the shell. The nacreous layer shows a similar zonation, with a similar rhythmicity (Fig. 6c). The organic walls between the prisms and the fibrous aragonite do not show a regular thickness. The surface of the organic walls is smooth within the prisms, but is irregular with thin extensions in the fibrous aragonite. Regular growth lines are also visible at higher magnification in the fibrous aragonite (Fig. 6c).

Confocal and IR maps. a Confocal microscope image of the inner surface of the shell, showing the first stage of the fibrous layer (FA) on the thick organic envelopes of the calcite prismatic layer (P). N nacreous layer. b Confocal microscope image of the polished vertical section showing the growth lines in the calcite prismatic layer (P), the nacreous layer (N), and the fibrous aragonite (FA). c Detail of the same zone, showing the growth layer in the nacre (N), the prismatic layer (P), and the fibrous aragonite (FA). d Polished vertical surface showing the locations of the IR maps (e–h). e IR map showing the composition of the nacreous layer (N) and fibrous aragonite. w organic wall. f IR map indicative of α-helix structures in the nacreous and fibrous layers. g IR map at 1,656 cm−1 assigned to the α-helix. h IR map related to C=O of asparagine. i, j Polished vertical surface showing the locations of the IR maps (k–n). k IR map showing the calcite composition of the prisms (P) and the composition of the nacre (N) and fibrous layers (FA). l Amide II band maps at 1,542 cm−1 showing the organic thick wall (w). m Amide II band map at 1,558 cm−1: the prismatic layer has relatively large transverse bands. n Map at the 1,661-cm−1 band (amide I) showing heterogeneities in both nacre and prisms
Two series of IR maps of the three layers were made (Fig. 6d–n), one of them including the interprismatic organic wall. The first set of maps (Fig. 6d–h) show differences in the intensities at several wavelengths, and in the map at the 1,489-cm−1 band (Fig. 6e), usually assigned to aragonite. This band is also characteristic of C–N asymmetric stretching and N–H group deformation. Thus, some parts of the organic wall between the fibrous aragonite and the prisms produce a high intensity at the 1,489-cm−1 band. The amide II response at 1,548 cm−1 (Fig. 6f) is indicative of α-helix structures and this map is similar to that at 1,489 cm−1, with high intensities in the organic wall. Discontinuities in the organic wall may be due to differences in composition, but may also reflect differences in thickness. The amide I response at 1,656 cm−1 is also assigned to the α-helix. This map (Fig. 6g) shows a low intensity in the nacreous layer, with highest intensities in the prismatic layer. The amide I response at the 1,677-cm−1 band (Fig. 6h) may be related to the presence of C=O of asparagine. This map does not show strong differences between the organic wall and the prismatic layer.
The second set of maps includes an interprismatic organic wall on a smaller region (Fig. 6i–n). The 1,429-cm−1 band is assigned to calcite for the mineral component. In organic components, the carboxylate symmetric stretching occurs between 1,429 and 1,432 cm−1, so the map at the 1,429-cm−1 band (Fig. 6k) includes both the mineral and the organic components. The organic wall has a higher intensity response, and a faint banding can be seen in the calcite layer. The amide II band maps (1,542 and 1,558 cm−1) are similar (Figs 6i,m): they show that the concentrations of contributing components are lower in the thick interprismatic organic wall in organic layer between the prismatic and nacreous layers. Moreover, the fibrous aragonite has a broadly similar response to the nacre, whilst the prismatic layer has a series of relatively large transverse bands with variable intensities. The map at the 1,661-cm−1 band (amide I) shows some differences: the nacre and fibrous aragonite have similar intensities, but with a lower content than in the prismatic layer. No bands are visible in the prismatic layer, whereas the interprismatic wall has a higher content intensity than the organic layer between nacre and prisms.
Thus, both optical confocal microscope images and IR maps confirm the composition of the fibrous layer.
NanoSIMS maps
NanoSIMS maps were focused on “organic” components in the transition zone between prisms and nacre in two mapped areas (Fig. 7a–c,d–f). The 12C maps clearly show the interlamellar organic layers of the nacre, and the composite structure of the organic walls between the calcite and fibrous aragonite (Fig. 7a,d). The interprismatic wall and the thick organic layer between prisms and nacre are continuous, and are composed of three layers. Growth lines in both layers are present in the 14N maps, where the organic walls are enriched in 14N (Fig. 7b,e). The 32S maps do not show significant differences between the layers, with the organic interlamellar membranes of the nacre being the most pronounced (Fig. 7c,f). Detailed images of the fibrous layer show faint growth lines (Fig. 7d,e), the thicknesses of which are similar to those of the nacreous layer. However, growth lines at the top of the prisms are not visible (Fig. 7d,e), and the three layers of the interprismatic wall are also not so prominent.

NanoSIMS (a–f) and XANES (g-k). a NanoSIMS 12C, showing the layered structure of the nacreous layer (nac), the fibrous aragonite (FA) and the calcite prisms (cp). b 14N map showing the similar content in the nacreous and prismatic layers, and the high content of the organic wall. c 32S map showing the low content of the organic wall. d 12C map showing the low content of the fibrous aragonite (FA) and the high content of the organic envelope of the prisms. e 14N map showing the low content of the fibrous aragonite (FA). f 32S map showing the low content of the fibrous aragonite, and the layered structure of the nacreous layer. g XANES map showing the similar Na content of the nacre and fibrous aragonite. h map showing the relatively higher Mg content of the calcite prisms. i map showing the similar Sr content in the three layers. j S amino acid XANES map showing the high contents of the organic wall (in red); E=2.473 keV. k SO4 XANES maps showing the high contents at the end of the calcite prisms; E=2.482 keV. For the NanoSIMS images, dark blue colours indicate relatively lower contents. For the XANES maps, the colour scale corresponds to the visible light spectrum, where “blue” represents low intensity, and red to high intensity
A potential caveat from these maps is that N is measured as the 12C14N− ion. This means that detection of N can only be made from regions containing C. However, because the target consists primarily of CaCO3 with embedded organics, there are no regions in which C is not available to generate the 12C14N− ion. Although the yield of 12C14N− ions can be sensitive to the chemical properties of the matrix from which they are sputtered, the NanoSIMS images obtained provide a strong indication that N is primarily associated with the organic membranes that separate the calcite prisms and the individual layers in the nacre.
Thus, NanoSIMS maps show the layered structure of the fibrous layer between the nacre and prisms. Moreover, growth lines are visible in this layer, and there is the indication that the thick organic envelopes have a composite structure.
Micro-XANES maps—sulphur speciation
The microfluorescence maps (Fig. 7g–k) across the nacre–calcite prisms boundary show similar results and correlate well with the electron microprobe maps. Relatively higher Na contents occur in the nacreous layer (Fig. 7g), and a higher Mg content is present in the calcite prisms (Fig. 7h); Sr is at similar concentrations in the nacre, prisms, and fibrous layer (Fig. 7i).
The most interesting feature of the XANES maps can be seen in the maps for sulphur (Fig. 7j,k). The high spectral resolution around the sulphur K-edge obtained using the micro-XANES setup of ID21 at ESRF enables different chemical states of sulphur to be mapped on the basis of minor differences in energy levels [2, 3], and at a high spatial resolution. In this study, the map at energy (E = 2.4735 keV) corresponding to the distribution of S from the amino acids cysteine and methionine shows that S amino acid content is higher in the nacreous layer than in the calcite prisms (Fig. 7j). However, the highest concentration of cysteine/methionine corresponds to the organic walls between the calcite prisms, and between the prisms and the fibrous aragonite (Fig. 7j). The map for organic sulphate (E = 2.4824 keV) shows a different pattern (Fig. 7k), with the top of the prisms having the highest concentrations; the nacreous layer is relatively homogeneous in terms of organic sulphate content, and has a slightly lower content of organic sulphate than the prisms. Within both the prisms and the nacreous layer, growth lines are clearly visible. However, of note is that the organic walls do not show any enrichment in organic SO4.
Thus, the total S contents of the fibrous layer and the nacre are similar, and are different from those of the calcite prisms.
Discussion and conclusion
Our microstructural, mineralogical, and chemical study of the nacre–prisms boundary in the shells of Pinctada margaritifera shows that this boundary is not an abrupt transition. This irregular boundary layer is of such small size that our study required the employment of a combination of several in situ, high-resolution techniques to investigate the structure and chemical composition of this region. Our results show that the boundary layer is in fact a distinct zone, relatively variable, and is neither nacre, nor prismatic calcite. The mineralogy and chemical composition of this zone are indicative of a fibrous layer that produces topographic structures, and evidence of growth lines. Results from confocal microscopy, XANES, and IR maps show that the organic components of this layer are similar to those of the nacreous layer. However, the changes in the biomineralization process are not restricted to this fibrous layer. IR, XANES, NanoSIMS, and some element maps also show that the composition of the final growth stage of the prismatic layer is different from that of the main part of this layer. The three-layered structure of the organic envelope of the calcite prisms was already visible in XANES maps [2]: these envelopes show high concentrations of sulphur amino acids, with a heterogenous pattern between the median and the lateral zones.
In Pinctada radiata, the tall columnar cells lining the outer surface of the outer mantle fold are responsible for the production of the calcite prismatic layer [9]. Similar cells have been described in Pinctada margaritifera [10]. Jabbour-Zahab et al. [10] also described two areas in the mantle epithelium that are very rich in acidic glycoproteins, and are probably related to shell secretion. Moreover, an intermediate area, poor in acidic secretion, may be involved in the transition layer between aragonite and calcite.
We have been able to identify at least three stages of growth from the prismatic structure to the nacreous layer. Firstly, in the terminal stage of the prism, a change can be seen in the chemical and biochemical composition of the last growth increments, although no change is visible in the microstructure or in the mineralogy. Secondly, there is a change in the mineralogy and microstructure within the overall growth process, and the chemical composition becomes similar to that of the nacreous layer. Finally, the microstructure becomes the nacreous layer, with the formation of regular tablets. Thus, there is good correspondence between the histological areas identified by Jabbour-Zahad et al. [10] and the three-step biomineralization process outlined here.
The nacreous layer is the most studied of the shell layers. Several proteins have been characterized and cloned in several species of Pinctada [11–14]. In contrast, fewer studies have been undertaken on the prismatic layer [15], because this layer is not thought be involved in pearl formation. However, Miyashita et al. [16] have shown that an identical carbonic anhydrase contributes to the secretion of the nacreous and prismatic layers in Pinctada fucata. Takeushi and Endo [17] have examined the possible role of six shell matrix proteins in Pinctada fucata. They found that two (MSI60 and N16) are present in the dorsal zone of the mantle, three (MSI31, prismalin 14, and aspein) are present in the ventral zone, whereas nacrein is present in both ventral and dorsal zones. In addition to the cloning of proteins to obtain sequence data, they used in situ hybridization to display their localization in the mantle. They suggest that the first group of proteins is involved in the secretion of the nacreous layer, and the second group is involved in the secretion of the calcite prismatic layer. The ubiquitous nacrein is said to be a carbonate-producing enzyme.
Because of commercial implications, many papers have been devoted to the growth of Pinctada; however, most are concerned with the length of the shell (e.g. [18]), not the thickness, so it is difficult to estimate the duration of the secretion of the transition from prisms to nacre. However, the daily rate of deposition of the nacreous layer in Pinctada margaritifera ranges from 1.3 to 5.9 μm (average 3.45 μm) [19]. As the maximum thickness of the fibrous layer is 20 μm, we suggest that the changes in the secretion process last for approximately 6 days.
Detailed comparisons of the growth strategies of the nacreous and prismatic layers have been reported in the Pteriomorpha bivalve Atrina rigida [20]. However, despite the fact that Atrina and Pinctada are taxonomically related (both are Pteriomorpha), the compositions of the intraprismatic soluble organic matrices of the calcite prisms of Pinnoidea and Pterioidea are very different. The main part of the intraprismatic soluble organic matrix of Pinna (closely related to Atrina) is composed of acidic and sulphated sugars, whereas that of Pinctada is mainly composed of acidic proteins [21]. Another difference is the calcite prisms: monocrystalline in Pinna and Atrina, they are polycrystalline in Pinctada. So the models based on Atrina are not fully appropriate to Pinctada.
From a practical and economical viewpoint, improvement of our understanding of the transition process between the prismatic and nacreous layers of the Pinctada shells is important for the pearl industry. In pearl production sites, the success of grafting operations, by which a nacreous layer will develop, relies first on the cutting of a mantle epithelium fragment that is deposited onto the “nucleus”, a mineralized sphere previously introduced within the mollusc body. Proper cutting of this epithelium fragment is essential to the quality of the subsequent pearl. To date, selection of the best place to cut the epithelium fragment is empirical. A better understanding of the secretory mechanism that results in the progressive formation of the true nacreous layer may contribute to improving the efficiency of the grafting process.
References
Mutvei H, Dauphin Y, Cuif JP (1985) Bull Mus Batl Hist Nat Paris 4 Ser 7 A 1:73–91
Dauphin Y, Cuif JP, Doucet J, Salomé M, Susini J, Williams CT (2003) J Struct Biol 142:272–280
Dauphin Y, Cuif JP, Salomé M, Susini J (2005) Am Mineral 90:1748–1758
Wada K (1957) Bull Natl Pearl Res Lab 2:86–93
Jackson DJ, McDougall C, Green K, Simpson F, Wörheide G, Degnan BM (2006) BMC Biol 4:40. DOI 10.1 186/1741-7007-4-4
Cox RL (1969) In: More RC (ed) Treatise on invertebrate paleontology. Geological Society of America and University of Kansas, Boulder
Milliman JD (1974) Marine carbonates. Part 1. Springer, Berlin Heidelberg New York
Dauphin Y, Gautret P, Cuif JP (1996) Bull Soc Geol Fr 167:247–256
Nakahara H, Bevelander G (1971) Calc Tiss Res 7:31–45
Jabbour-Zahab R, Chagot D, Blanc F, Grizel H (1992) Aquat Living Res 5:287–298
Miyamoto H, Miyashita T, Okushima M, Nakano S, Morita T, Matsushiro A (1996) Proc Nat Acad Sci USA 93:9657–9660
Samata T, Hayashi N, Kono M, Hasegawa K, Horita C, Akera S (1999) FEBS Lett 462:225–229
Kono M, Hayasi N, Samata T (2000) Biochem Biophys Res Commun 269:213–218
Miyashita T, Takagi R, Okushima M, Nakano S, Miyamoto H, Nishikawa E, Matsushiro A (2000) Mar Biotechnol 2:409–418
Suzuki M, Murayama E, Inoue H, Ozaki N, Tohse H, Kogure T, Nagasawa H (2004) Biochem J 382:205–213
Miyashita T, Takagi R, Miyamoto H, Matsushiro A (2002) Veliger 45:205–255
Takeushi T, Endo K (2006) Mar Biotechnol 8:52–61
Pouvreau S, Prasil V (2001) Aquat Living Res 14:155–163
Mavuti KM, Kimani EN, Mukiama T (2005) J Mar Sci 27:567–575
Nudelman F, Chen HH, Goldberg HA, Weiner S, Addadi L (2007) Faraday Discuss 136:9–25
Dauphin Y (2003) J Biol Chem 278:15168–15177
Acknowledgements
This work has been made possible thanks to scientific support from the ESF under the EUROCORES Programme EuroMinScI (BioCalc project), through contract no. ERAS-CT-2003-980409 of the European Commission, DG Research, FP6, financial support of the French ANR through contract ANR-06-BLAN-0233, ESRF grants CH1569 and EC24, and EU-IHP Programme (SYNTHESYS) for Y.D. and J.P.C to visit the NHM, London.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dauphin, Y., Ball, A.D., Cotte, M. et al. Structure and composition of the nacre–prisms transition in the shell of Pinctada margaritifera (Mollusca, Bivalvia). Anal Bioanal Chem 390, 1659–1669 (2008). https://doi.org/10.1007/s00216-008-1860-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-1860-z