
Abstract
Due to the production and use of a multitude of chemicals in modern society, waters, sediments, soils and biota may be contaminated with numerous known and unknown chemicals that may cause adverse effects on ecosystems and human health. Effect-directed analysis (EDA), combining biotesting, fractionation and chemical analysis, helps to identify hazardous compounds in complex environmental mixtures. Confirmation of tentatively identified toxicants will help to avoid artefacts and to establish reliable cause–effect relationships. A tiered approach to confirmation is suggested in the present paper. The first tier focuses on the analytical confirmation of tentatively identified structures. If straightforward confirmation with neat standards for GC–MS or LC–MS is not available, it is suggested that a lines-of-evidence approach is used that combines spectral library information with computer-based structure generation and prediction of retention behaviour in different chromatographic systems using quantitative structure–retention relationships (QSRR). In the second tier, the identified toxicants need to be confirmed as being the cause of the measured effects. Candidate components of toxic fractions may be selected based, for example, on structural alerts. Quantitative effect confirmation is based on joint effect models. Joint effect prediction on the basis of full concentration–response plots and careful selection of the appropriate model are suggested as a means to improve confirmation quality. Confirmation according to the Toxicity Identification Evaluation (TIE) concept of the US EPA and novel tools of hazard identification help to confirm the relevance of identified compounds to populations and communities under realistic exposure conditions. Promising tools include bioavailability-directed extraction and dosing techniques, biomarker approaches and the concept of pollution-induced community tolerance (PICT).

Toxicity confirmation in EDA as a tiered approach
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Due to the large-scale production and use of an increasing diversity of chemicals in modern society, surface and ground waters, sediments, soils, biota tissues, foods and feedstock may be contaminated with a multitude of known and unknown chemicals. Some of these chemicals may pose hazards to organisms including humans, as indicated by effects observed, for example, in bioassays (both in vivo and in vitro). It is often crucial to identify the components responsible for adverse effects in toxic samples in order to be able to identify sources as well as assess and mitigate risks. Effect-directed analysis (EDA) and toxicity identification evaluation (TIE), which both combine biological and chemical analysis with physicochemical manipulation and fractionation techniques, have been shown to allow for toxicant identification in many matrices and for many toxicological endpoints [1–6]. While TIE originates from effluent control in a regulatory context in the US, EDA is a more scientific approach developed by analytical chemists to identify unknown hazardous compounds in various environmental or technical matrices. TIE is based exclusively on in vivo testing, while EDA is applied to both in vitro and in vivo tests in order to detect active fractions and compounds. EDA is not restricted to identifying the cause(s) of acute toxicity (e.g. in effluents), but also aims to identify potentially hazardous compounds in the environment, even if the concentrations present should not cause acute effects. Thus, extraction and preconcentration procedures as well as the analysis of sensitive sublethal biochemical in vitro responses are important tools in EDA.
TIE is based on guidelines by the United States Environmental Protection Agency (US EPA) [7–9] that explicitly demand toxicity confirmation as the final step in this procedure. This step is intended to confirm the findings of toxicity characterisation and identification. No generally agreed guidelines are available for EDA. Thus, what is meant by “confirmation” is less clearly defined. We define confirmation in EDA as providing evidence in several steps. Firstly, that suggested chemical structures derived from analytical procedures correctly match the compound actually present in the sample; secondly, that the compounds substantially contribute to the measured effect of the mixture; and thirdly, how much of the effect may be explained by the identified compounds and (more importantly) how much is not explained are evaluated. While confirmation seemed to play a minor role in practical EDA and TIE in the 1980s and 1990s, more recently the awareness of research needs in this field has increased, particularly in tandem with progress in mixture toxicity prediction [10, 11]. The present paper aims to briefly discuss the state of the art in confirmation in EDA, including analytical and effect confirmation, in order to discuss the lessons that can be learned from TIE confirmation approaches and to identify open questions and research needs in EDA confirmation.
Confirmation as a tiered approach
As a starting point for confirmation, we consider a list of tentatively identified compounds in a biologically active fraction based on a library search of mass spectra. Such a list provides a first idea of possible causes of measured effects. However, there are good reasons to be sceptical that the compounds tentatively identified in the toxic fraction, e.g. by gas chromatography with mass-selective detection (GC/MS) along with a search in the spectral library of the National Institute of Standards and Technology (NIST), actually explain the measured effects:
-
1)
Particularly with respect to isomers, which might have very different toxicological activities, even a good agreement with NIST spectra does not provide proof of the correct identification of the individual structure.
-
2)
The toxicant actually responsible might co-elute with other analytes, or it might be below the detection limit or be undetectable using the selected analytical method (e.g. thermolabile compounds in GC).
-
3)
Unknown and interfering modes of action and unpredictable mixture effects, including toxicity masking, might confuse and lead to the wrong results even when individual toxicity data on identified compounds are available.
-
4)
Different bioavailabilities may occur in real samples and artificial solutions of individual compounds and mixtures.
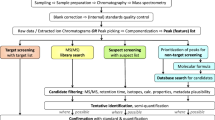
Thus, it is obvious that consistent concepts and approaches to confirmation are crucial to reliable toxicant identification. Confirmation (Fig. 1) may be seen as a tiered approach, where the first step should be the analytical confirmation of the suggestions resulting from a library search. When we are sure about the chemical structure, qualitative and quantitative toxicity confirmation in vitro or in vivo, based on the endpoint used in the EDA study, should provide an estimate of whether the compound can explain the measured effects, and to what extent [9]. If we are only interested in the in vitro or in vivo effects themselves, we can then consider the toxicant(s) as being confirmed. However, we often use EDA approaches in ecological risk assessment, and thus we need to identify those chemicals that pose a risk to the community or ecosystem [12]. Since causal analysis on the basis of in situ effects is often not very promising, we use in vitro or in vivo bioassay-based EDA to answer this question. However, we should be aware that toxicity in simple laboratory test systems is not a direct predictor for in situ hazards. Thus, additional confirmation steps that prove the potential hazards of the identified toxicants at the community level under realistic exposure conditions are required. First ideas are presented in this paper.

Toxicity confirmation in EDA as a tiered approach
Analytical confirmation
Analytical confirmation starting from compounds tentatively identified on the basis of mass spectra may be seen in many cases to be a stepwise approach leading to increased evidence rather than a clear yes/no decision. The additional evidence we can gain at every step depends on the compound, the amount available in the sample, the availability of standards and the resolution of the separation step performed prior to identification and confirmation. Numerous compounds in a fraction (insufficient fractionation), co-eluting compounds and compounds with many possible isomers with similar physicochemical properties [e.g. alkylated polycyclic aromatic hydrocarbons (PAHs), alkylphenols] significantly reduce the additional evidence that can be obtained from the individual steps of analytical confirmation. Thus, extensive fractionation and high chromatographic resolution prior to mass spectral recording are major prerequisites for successful confirmation. These may include, for example, multistep fractionation procedures [13], preparative capillary gas chromatography [14] and two-dimensional GC techniques [15].
Generally, the agreement of retention times and mass spectra with those of neat standards and structural elucidation by nuclear magnetic resonance (NMR) spectroscopy are considered to provide sufficient evidence for full confirmation. Unfortunately, there are currently no available standards for many compounds, and even full standard availability would not allow a straightforward confirmation based on retention times and mass spectra for some compounds with co-eluting isomers (e.g. alkylated PAHs). NMR has already been performed in EDA studies [16–18]. However, NMR is difficult to perform for many environmental trace contaminants because of the high amounts and the purities of the analytes required. For example, 27 kg of the adsorbent blue rayon had to be deposited in river water to collect sufficient amounts of benzotriazole-type mutagens for structural elucidation and confirmation with NMR [16].
The first step in compound identification for thermally stable compounds that are analyzable by GC/MS is, in most cases, a comparison with spectral libraries in order to select matches with good fits. Compounds that are identified based on this procedure are generally referred to as “tentatively identified”. That means that we have a well-founded idea of possible structures that fit the spectral information we obtained for the specific analyte. However, without further lines of evidence it is generally impossible to reliably associate spectra with one specific compound.
A further line of evidence is a close match of retention times or indices with published data for authentic standards run under similar conditions [19]. Compounds with mass spectra and retention times or indices that closely match literature or library data may be referred to as having “confident structural assignment” [20]. The analysis of mass spectra together with retention indices using the Automated Mass Spectral Deconvolution and Identification System (AMDIS) [21] is a powerful tool that supports this procedure. Accurate masses determined with high-resolution mass spectrometry provide exact elemental compositions of analytes and thus are an important tool used to further enhance evidence and to reduce the number of possible structures.
However, we should also be aware of the major shortcomings of the library approach: compounds that are not included in the library cannot be correctly identified. The library of NIST [22] is one of the most frequently used libraries and includes about 190,000 mass spectra of 163,000 compounds. Although this is a huge number, it covers only a small portion of all known or possible chemicals. Thus, a more generic approach that provides the full set of possible chemical structures with reasonable fits to the recorded mass spectrum would be helpful as a starting point for the identification and confirmation of unknowns. One tool that can be used to do this is the MOLGEN software [23–25], which generates the full set of mathematically-possible structures that fit well to a given molecular mass identified using mass spectrometry. The difference between a structure generator such as MOLGEN and spectral libraries can be demonstrated with few simple numbers. For a molecular mass of 150, a total of 376 molecules are available in spectral libraries. The Beilstein database of known compounds contains 5300 molecules, while MOLGEN is able to generate 615,977,591 molecules with this molecular mass [24]. Thus, a library search for this molecular mass is based on 6 × 10−5% of all possible compounds and 7% of all known structures. For higher molecular weights the number of possible structures increases exponentially, while the numbers of known compounds and compounds included in mass spectral libraries remain almost constant. Of course, not all “mathematically possible” molecules (i.e. molecules generated on the basis of standard valences of the atoms contributing to a molecule) are energetically or kinetically stable and thus likely to be found. However, these numbers help to illustrate the challenges of analytical identification and confirmation.
A significant reduction in the number of possible compounds can be achieved by recording high-resolution mass spectra that provide exact masses and thus reliable data on the elemental composition of the analyte. The MOLGEN-MS software combines structure generation based on the summation formula with mass spectral “classifiers” that allow substructures to be identified as being probable or improbable based on mass spectral fragmentation patterns. These classifiers can be included in structure generation via the use of good and bad lists [26]. The effectiveness of this software is largely dependent on the “classifiers” available, but when sufficient classifiers exist, the program can be very effective in reducing the number of possible structures. Unfortunately, the model still exhibits several inconsistencies that, at present, prevent the full exploitation of this promising tool. The advancement of MOLGEN or the development of new tools for structure generation basis on mass spectral information is a high-priority need with respect to structure elucidation and confirmation in EDA and other applications.
Retention indices are a highly important tool for further reducing the number of possible structures as derived from mass spectra. However, retention indices are only available for a limited number of compounds. Thus, the estimation of these values using quantitative structure–retention relationships (QSRR) will become increasingly important. These methods may help to substantially reduce the number of possible isomers that fit the chromatographic and spectral information of the unknown. Simple plausibility checks based on the correlation of boiling points with retention indices in combination with estimated boiling points for the tentatively identified structures may help to exclude compounds with physicochemical properties that do not fit the observed chromatographic behaviour. An example of such a correlation of boiling points with the Lee retention index (RI) with reasonably narrow prediction bands was recently presented [27]. Using this correlation, boiling points (BP) were predicted on the basis of RI (BPRI) for 370 compounds from 20 chemical classes. 95% of the experimental boiling points were within the range (BPRI−10) to (BPRI+50). Another promising approach is the prediction of retention times and indices on the basis of linear solvation free energy relationships (LSER) according to the Abraham model [28, 29]. As opposed to boiling point, which is a property of the analyte alone, the Abraham equation is based on fundamental types of intermolecular interactions between the bulk phases (in chromatography the stationary and the mobile phase) and the partitioning analytes, considering hydrogen bond acidity, hydrogen bond basicity, dipolarity, excess molar refraction and molar volume. An excellent review of chemical interpretation and the use of LSER was published recently [30]. This approach has the potential to provide a powerful tool to check tentatively identified structures for plausibility, if Abraham parameters can be reliably estimated from chemical structure. Multidimensional predictions could help to further reduce the number of possible structures that fit the analytical results if several stationary phases with substantially different but known Abraham descriptors were to be used. To date there is only one model available for the prediction of Abraham descriptors [31]. Recent investigations have demonstrated that, at present, this model still shows several inconsistencies and the domain is restricted to relatively simple compounds with few functionalities [32]. An extension of this domain to compounds with two or more functional groups and an improvement in descriptor prediction from chemical structure could help to make the Abraham model a valuable tool for the identification and confirmation of toxicant structures in EDA.
For compounds with rather nonspecific mass spectra that are similar despite different basic structures, such as PAHs and related compounds, additional information such as ultraviolet (UV) and infrared (IR) absorption spectra and capacity factors on different HPLC columns can significantly enhance the evidence of confirmation. One example is the identification of 11H-indeno[2,1,7-cde]pyrene in a highly mutagenic fraction of a sediment extract in the Neckar basin [33]. The mass spectrum, which had a molecular ion of 264 and a high intensity of the M-1 ion, suggested a PAH with a parent mass of 252 and an additional CH2 group bridging two aromatic carbons. This still allows several isomers, since parent PAHs with M = 252 include more than 20 isomers. However, the UV spectrum clearly indicated a benzo[e]pyrene derivative, which leaves only one possible structure containing a benzo[e]pyrene structure and a CH2 bridge if rings with three or four carbon atoms are excluded.
A strong enhancement of the evidence can be achieved if pure standards that are commercially available or that have been synthesized and carefully characterised by NMR can be applied. However, it should be stressed that the agreement, for example, of the mass spectrum and a retention index of an analyte with those of a standard does not mean full confirmation in every case. Again, methylated PAHs serve as an example of different compounds with very similar retention indices and mass spectra.
When identifying thermally labile compounds that are not suitable for analysis using GC/MS, the first step of tentative identification may already be very difficult because of the absence of extensive libraries for LC/MS techniques. The application of orthogonal-acceleration time-of-flight (TOF) MS, which permits the recording of the accurate mass and thus elemental composition, as well as hybrid instruments combining quadrupole and TOF-MS (LC–Q-TOF-MS), provides new lines of evidence for a specific structure [34]. Tandem mass spectrometric approaches (LC–Q-TOF-MS/MS) have been shown to be powerful tools for the identification of unknowns [35, 36]. A generally agreed concept for the confirmation of structures of unknowns that have been tentatively identified with LC–MS techniques is not yet available. For target analysis of polar compounds with LC–MS, a confirmation concept based on the principle of the number of identification points was proposed by the European Commission for food contaminants [37]. Increasing numbers of precursor and product ions in LC–MSn analysis give increasing numbers of identification points, indicating greater confidence in correct compound identification.
LC–MS–MS in its various forms is a powerful technique for the identification and confirmation of unknowns. We should differentiate between two types of “unknowns”; the so-called “suspected unknowns”, such as pesticide and/or pharmaceutical degradation and phototransformation products in the environment, and the “real unknowns” or “unknown unknowns”.
One very powerful tool is the combined approach of time-of-flight mass spectrometry (TOF-MS) and ion trap (IT) MS instruments, as well as hybrids combining quadrupole and time-of-flight techniques (QqTOF-MS) or quadrupole and linear ion trap (QqLIT-MS), which allow either accurate mass determinations or fragmentation patterns to be obtained based on MSn experiments, therefore providing complementary structural information. Combinations of ultrahigh-performance liquid chromatography (UPLC)–QqTOF-MS and LC–IT/MSn or LC–QqLIT-MS in the same basic format can be applied as a universal generic strategy to find, characterise and confirm unknown compounds in environmental and food analysis. The availability of the full precursor or product mass spectra throughout each HPLC or UPLC chromatogram and either accurate mass measurements or multiple stages of mass spectrometry will provide qualitative information that can be used to ascertain whether metabolites or any other compounds are present in the sample. The knowledge of the exact analyte masses allows the determination of possible molecular formulae and chemical structures for the suspected metabolites, while the sequential fragmentations provide a better understanding of the fragmentation patterns. It seems feasible that this approach could be useful for providing information on metabolites while simultaneously achieving their quantification.
Examples of the identification of so-called “suspected unknowns” through the combined use of both techniques have recently been reported in the literature, such as the identification of four new phototransformation products of enalapril [38]. Accurate mass measurements recorded on a hybrid quadrupole–time-of-flight (QqToF) instrument in MS/MS mode allowed the elemental compositions of the molecular ions of the transformation products (346 Da: C19H26N2O4; 207 Da: C12H17NO2; 304 Da: C17H24N2O3) to be proposed, as well as those of their fragment ions. Based on these complementary data sets from the two distinct mass spectrometric instruments, plausible structures could be postulated.
Another recent study combining ITMS and QqTOF-MS [39] showed that fenthion in orange orchards is mainly transformed to its sulfoxide. From the residues detected, fenthion sulfone was found to be constantly present, although in low quantities. Fenoxon, fenoxon sulfoxide and fenoxon sulfone can always be detected in low quantities following rain events (even lower than those of fenthion sulfone). This represents an important outcome, taking into account that all of these reported metabolites are also toxic.
The second class of unknowns is the so-called “real unknowns” and/or “unknown unknowns”. Basically, these compounds are detected by the accurate mass measurements of QqTOF when analyzing other target compounds, since TOF analyzers offer full mass spectra with accurate masses, and consequently the ability to look for other analytes in a specific target analysis. Two examples are reported here. In the first case, when selected estrogens such as estrone, estradiol and ethynyl estradiol were analysed in the Q-TOF-MS mode, Q-TOF allowed the identification of non-target and/or suspected compounds such as the phytoestrogens daidzein, genistein and biochanin [40]. Another recent paper deals with the use of UPLC–QqTOF-MS to identify residues of the pesticide imazalil in complex pear extracts [41]. The non-target pesticides carbendazim and ethoxyquin were successfully identified and confirmed because of the accurate mass determination of their protonated molecules and major fragments in the product ion mass spectra. The main product ions of the three non-target pesticides present together with imazalil residues were coincident with those identified by triple quadrupole mass spectrometry, indicating unequivocal identification of these unknown pesticides. These examples of pesticide residues highlight the power of this technique to identify non-target molecules without the use of standards beforehand. Published literature on the subject, chemical databases and websites combined with the QqTOF results yield a very useful tool for unambiguously identifying compounds. The main problem with this modus operandis is the lack of libraries that enable a possible structure to be searched for given a particular elemental composition within the equipment software.
In summary, the analytical confirmation of trace contaminants detected in EDA studies is still a challenging task. There are no generally agreed guidelines, and the effort required depends upon the sample complexity and composition as well as on the individual component. However, the following suggestions and research needs may be derived from experience so far:
-
1)
Extensive fractionation and high chromatographic resolution prior to recording the mass spectra are a major prerequisite for successful confirmation.
-
2)
Where no neat standards are available, analytical confirmation is a lines-of-evidence approach rather than a yes-or-no decision. Major lines of evidence are mass spectra and GC retention indices for volatile compounds and accurate mass spectra obtained by QqTOF MS/MS for nonvolatiles. Additional lines may be provided by HPLC capacity factors on different columns and UV and fluorescence spectra.
-
3)
At present, the comparison of measured chromatographic and spectrometric data with respective library data is a common approach. However, we should be aware that libraries do not contain all possible compounds and thus may fail to identify unknowns. Reliable prediction of chromatographic and spectrometric data from tentatively identified structures and automated comparison with measured data may overcome this shortcoming and open completely new windows to the successful identification and confirmation of unknowns.
-
4)
For nonvolatile compounds, first approaches to using LC–MS techniques for structure elucidation and confirmation are available. Confirmation can be achieved based on LC–QqTOF/MS and LC–MSn techniques with neat standards, by considering several precursor and product ions together with accurate mass measurements that allow toxicant identification.
-
5)
The challenge of identifying and confirming new toxicants in the environment necessitates common and generally accessible databases of chromatographic and spectrometric data for newly identified key toxicants generated under agreed standard conditions to allow for the exchange of relevant information. An attempt to create such a database is currently underway in the European Integrated Project MODELKEY [12].
Effect confirmation in EDA
While analytical confirmation aims to ensure that chemical structures in toxic fractions are correctly identified, effect confirmation aims to provide evidence that the identified compounds are actually responsible for the measured effects. Quantitative structure–activity relationships (QSAR) [42, 43] and structural alerts [44] are valuable tools for selecting potentially active compounds from identified components in a toxic fraction. Although rarely applied, these tools may provide important lines of evidence, even in the case where no neat standards are available for performing biotests. Within the last few years, substantial progress has been made in the prediction of specific effects and excess toxicity (more than narcotic effects) from chemical structure. Examples are the identification of excess toxicity in Daphnia magna [44], estrogenicity [43], binding to the androgen receptor [45], toxicity of arylhydrocarbon-receptor (AhR) binding compounds [46], as well as mutagenicity and carcinogenicity [42, 47, 48]. Particularly in cases where the final fractions still contain numerous compounds, QSAR provides a promising tool for reducing and prioritising the number of candidate compounds.
All approaches to quantitative toxicity confirmation are either explicitly or implicitly based on relating individual toxicities to a reference model that predicts joint effects. In EDA and TIE almost all confirmation studies are based on the concept of concentration addition (CA). This concept assumes additivity of concentrations of different components of a mixture standardised to their effect concentrations at a defined effect level. This concept may be expressed by the formula
where c i are the concentrations of components 1 to i that cause a specific effect X (e.g. 50% mortality), and ECX i are the effect concentrations of the single compound exhibiting the same effect [49]. The basic assumption of this concept is a similar mode of action. The validity of this concept was shown, for example, for the algal toxicity of photosynthesis inhibitors and the bacterial toxicity of protonophoric uncouplers [50].
This concept is the basis for the toxic unit (TU) approach, which is commonly used for in vivo biotests, as well as the summation of Toxicity Equivalent Quantities (TEQs) or Induction Equivalent Quantities (IEQs), which are both used for in vitro assays. In the TU approach, concentrations are converted to TUs by division with the respective effect concentration at a specific effect level [51, 52]. The sum of the TUs should be equivalent to the TU of the mixture, which is simply the reciprocal of the effect dilution of the mixture at the same effect level.
In vitro TEQs and IEQs are used in the same way. They are expressed by multiplying the concentration of a compound in a mixture by toxicity or induction equivalency factors (TEFs and IEFs), also referred to as relative potencies (REPs). These factors standardise the potency of all compounds to the potency of a reference compound. TEQs and IEQs are added to get an estimate of the potency of the mixture. This concept is well established for dioxin-like activity. IEF/TEF values are available for most test systems, including induction of ethoxyresorufin-O-deethylase (EROD) in rainbow trout liver cell line RTL-W1 [53], in the fish hepatoma cell line PLHC-1 [54], in H4IIE rat hepatoma cell line [55, 56] and activation of the AhR in the chemical-activated luciferase expression (CALUX) assay [57, 58]. It should be stressed here that IEF/TEF values for the respective bioassay need to be used to obtain reliable mass balances, rather than the general TEF values as agreed by the World Health Organisation for human, fish and wildlife risk assessment [59]. IEF/TEF values for other in vitro effects are increasing. Examples are estrogenic potencies [60, 61], inhibition of gap-junctional intercellular communication as a parameter for tumor-promoting potency [62] and mutagenicity [63, 64].
While TUs, TEQs and IEQs are easy-to-use, straightforward approaches, we should keep in mind that they are based on several assumptions that have been summarised recently [65]: (i) concentration–response relationships are monotonic functions over a given concentration range to which a specific bioassay is applied, (ii) mixture effects follow the concept of concentration addition, assuming a similar mode of action and that all components contribute to mixture effects, and (iii) that the comparison between expectation and observation is based on a point estimate, suggesting that the combined effect assessment is valid for all response levels. The latter assumption holds if dose–response relationships are parallel.
In general, concentration–response relationships are monotonic functions as long as no problems in the exposure regime occur, e.g. by exceeding the limits of compound solubility or micelle formation [65]. Ambiguity may occur for example for assays based on enzyme induction measured as enzyme activity. In this case induction and inhibition may overlap, resulting in bell-shaped concentration–response relationships and fraction- or compound-dependent maximum induction levels (efficacy), as observed for EROD induction in rainbow trout liver cell line RTL-W1 [66, 67]. Other examples are the occurrence of cytotoxicity at high concentrations in complex mixtures or mixtures containing partial agonists with lower efficacies at inducing a response in a given assay. Effects exceeding those of the least potent partial agonist in a mixture cannot be computed [68]. IEQs may strongly depend on whether they are calculated based on EC50 values relative to individual maxima [55] or on fixed effect levels [66]. Although the concept of concentration addition (CA) is almost generally used in EDA confirmation, it should be stressed that it is valid only if all compounds exhibit similar modes of action [49].
In the case of dissimilarly acting compounds, statistically independent responses are expected. This type of mixture toxicity is best described by the model of independent action (IA), also referred to as response addition [65]. For a binary mixture, independent action means that component B attacks those organisms that survived compound A without interaction between both effects. The effect for the binary mixture is calculated according to the following formula:
In a more general formulation for n compounds, this means:
The predictability of mixture toxicity using the IA model has been shown recently for the joint algal toxicity of 16 pesticides selected for strictly dissimilar modes of action [69]. However, this finding could also be confirmed for a mixture of structurally heterogeneous priority pollutants with mostly unknown modes of action [70]. Depending on the effect level, CA overestimated mixture toxicity by a factor of 1.6 to 5.0, while IA gave a good prediction.
If in vivo tests based on lethality, inhibition of growth or reproduction are used for EDA of complex contaminated environmental samples, similar modes of action of all toxic components are quite improbable. Thus, the application of CA is likely to result in an overestimation of the toxicity caused by the identified toxicants. This was shown, for example, for diazinon and ammonia, which were identified by TIE to concurrently occur and affect invertebrates in municipal effluents [10]. With respect to the precautionary principle, overestimation and thus overprotection by a factor of 5 is not very problematic. However, overestimation by a factor of 5 in EDA confirmation means that we may conclude that we have explained 100% of the measured toxicity with the identified toxicants, whereas these toxicants are actually only responsible for 20% of the effects, leaving 80% unexplained.
One might argue that, as opposed to in vivo tests, in vitro assays primarily detect compounds with similar modes of action, in which case CA is more likely to be applicable. However, it was recently shown for combined effects of mycotoxin mixtures on human T cell function as a functional in vitro assay that joint effects could be predicted within their experimental uncertainty by IA [71]. In the case that similarly and dissimilarly acting toxicants occur in a mixture, joint models combining IA and CA for distinct compounds, respectively, may provide the best fits of mixture toxicity prediction [65].
The dependence of confirmation quality on the applied mixture toxicity model was recently studied for the algal toxicities of two sediment extracts [11]. To obtain a quantitative measure of the agreement between joint toxicity predictions for identified toxicants (Fig. 2, dashed and dotted lines) and the measured toxicities of extracts and artificial mixtures (solid lines), the authors introduced an index of confirmation quality (ICQ). The little arrow indicates the prediction according to the TU concept. An ICQ of 1 means full agreement of a predicted or measured effect of the identified toxicant mixture with the extract toxicity. The comparison of both sediment extracts resulted in quite different findings. While the toxicity of an artificial mixture of identified key toxicants at one of the sites (Bitterfeld) was well predicted using the concept of IA, the mixture based on the composition of sediment contamination at the other site (Brofjorden) was in good agreement with the CA prediction. This could be explained by very heterogeneous contamination in Bitterfeld compared with Brofjorden, where probably only PAHs with similar modes of action could be found. However, for unknown samples, their compositions and thus the correct joint effect models are simply not predictable. Thus, predicting using both models helps us to estimate the uncertainty due to model selection and provides predictions for the extreme cases of only similarly or only dissimilarly acting compounds.

Index of confirmation quality for effect levels of between 10 and 90% and two different sediments. a represents Bitterfeld (Germany) and b Brofjorden (Norway). The vertical line at 1 indicates the extract toxicity, which is used a reference and set to 1 for all effect levels. ICQ values are given for the measured toxicity of the synthetic mixture (SM, solid line), expectations of combined effects of identified toxicants according to concentration addition (CA, dotted line) and independent action (IA, dashed line). The horizontal dotted line indicates the 50% effect level mostly applied in toxicity assessment. The little arrow highlights the estimate based on the TU approach (modified after [11])
While mixture prediction using CA is based on effect concentrations and is possible for one selected effect level (e.g. EC50), the application of IA requires concentration–effect relationships for the mixture and the components. Wherever these data are available, predictions obtained using both models will help to identify a range of confirmation quality. Although this is less convenient than a single number, it provides an idea of the possible error in the prediction due to the selection of the wrong model, and it provides a valuable “quality control” check.
The next assumption for the applicability of the TU/TEQ approach, in addition to the validity of CA for joint effect prediction, is that the mixture toxicity prediction should be independent from the effect level under consideration. In most cases, mixture toxicity prediction or toxicity confirmation is done for a 50% effect level. However, the use of EC50 values is a rather arbitrary choice. It might be assumed that a generally applicable model for toxicity confirmation should provide similar results if we focus for example on a 20% effect level.
The analysis of the dependence of ICQ on effect levels for two sediment extracts indicated an increase in ICQ with increasing effect level, by almost a factor of 10. Thus, for both joint effect models the identified toxicants explain a significantly higher proportion of the activity if the assessment is based on EC90 rather than EC20. As a result, obviously many more compounds contribute to toxicity at low effect levels than at high effect levels.
Receptor-mediated in vitro effects triggered by binding to the aryl hydrocarbon receptor (AhR) or to the estrogen receptor (ER) seem to be typical examples of test systems that only detect compounds with similar modes of action and suggest CA as a reasonable concept for the prediction of mixture effects [61, 72]. This is in agreement with the extensive use of TEQ/IEQ in EDA [67, 73, 74]. However, as discussed above for the in vivo TU concept, there are good reasons for quantifying receptor-mediated relative potencies (REP) in vitro using functions rather than point estimates [75]. Similar to TUs, REPs may substantially depend upon the effect level under consideration. This is due to the differing efficacies and slopes of dose–response relationships frequently observed in receptor-mediated in vitro assays. Unfortunately, confirmation based on full concentration–response relationships has not prevailed thus far, as it is felt that this approach makes confirmation a somewhat cumbersome and laborious process [76]. In addition it demands full concentration–response information for individual compounds rather than effect concentrations or TEF values alone, as given in most papers and databases. Since every estimate of effect concentration (ECX) is based on concentration–response data, it can be assumed that all compounds for which ECX values exist should also have concentration–response relationship functions, although they are not available to the scientific public. Based on the understanding that different efficacies and slopes violate basic assumptions of the TEQ concept, Villeneuve et al. [76] suggested the use of ranges of relative potencies (REPs) at EC20, EC50 and EC80 (REP20–80) instead of EC50-based REPs. This might reduce the effort compared with the use of full concentration–response relationships. However, it does not solve the problem of a lack of data for individual compounds. Typical REP20–80 values presented for individual PCDD/F congeners in the CALUX assay ranged by a factor of two to four between highest and lowest REP [57], while for environmental samples this range could be even larger [76]. In general, EC50-based REPs are used for confirmation, while the effect levels used in EDA are mostly low enough to include fractions that do not exceed the 50% effect level. For CALUX, EC20-based REPs were consistently lower than EC50-based ones, by a factor of about 2 [57]. We should note that in EDA this may cause a deviation between calculated and measured values of at least a factor of two. Thus, it cannot be excluded that an apparent 100% confirmation could mask the fact that half of the actual potency has been ignored.
It was suggested that the CA concept could be used for toxicity confirmation only when there is a clear indication of similar modes of action [11]. However, even then the toxicants should be confirmed preferentially based on whole dose–response plots.
Although mutagenicity has been the focus of EDA since 1980 [77, 78], quantitative confirmation of mutagens has rarely been performed. In most cases, these studies ended up with lists of tentatively identified compounds in mutagenic fractions together with some qualitative information on the mutagenicity of each compound [79]. In those cases where efforts have been made to quantify the contributions of fractions or individual compounds to the total mutagenicity, this was based on the CA concept of applying ratios of compound concentrations to minimum mutagenic concentrations [80]. Unfortunately, to the best of our knowledge, no data are available that indicate that CA is a model that actually predicts mutagenicity in complex mixtures. Other than for receptor-based assays, dissimilar modes of action resulting in mutagenicity are likely to occur [81]. Concentration–response relationships of fractions and compounds may dramatically differ in terms of shapes, slopes and maximum values [82]. A recent study that systematically investigated the genotoxicity of PAHs in complex mixtures indicated an overestimation of mixture mutagenicity for 75% of the samples when predicted with CA [83]. The prediction of complex mixture mutagenicity from individual compound activities may be further impeded by the suppression of the activation and thus indirect mutagenic activity by mixture components [84]. However, direct mutagenicity may also be suppressed by other compounds [85]. The observation of substantially higher mutagenicity in fractions compared with the parent extract is a frequently observed phenomenon that might be explained by the sequential removal of mutagenicity-masking compounds [33].
Thus, although adding benzo[a]pyrene equivalents (BEQs) based on benzo[a]pyrene equivalency factors for individual mutagens [63] might provide a protective estimate for risk assessment, it is obvious that major assumptions for applying CA to mixture mutagenicity prediction are not met. The application of this concept in EDA studies is likely to result in an overestimation of the share of identified mutagenicity and to overlook substantial causes of mutagenicity. It may be concluded that, at the present state of knowledge, no quantitative approach is available for predicting mixture mutagenicity and the contribution of individual compounds to mixture toxicity with sufficient reliability to be used in EDA confirmation studies. At present, extensive fractionation, isolation and testing of mutagenic components might provide the most reliable way to identify and confirm mutagens in complex mixtures [33].
In summary, the general application of the TU or TEQ approach to effect confirmation in EDA without carefully proving that the approach is valid for each specific case results in inadequate modelling of the complexity of joint effects and may lead to substantial underestimation of the unexplained toxicity. The applicability of this concept is bound to similar modes of action that can only be assumed if specific mode-of-action tests (e.g. on receptor-mediated effects) are applied. In other cases, predicting the correct model for an unknown mixture is almost impossible. The following recommendations may be derived from present knowledge:
-
1)
The application of the TU/TEQ approach to confirmation studies should be restricted to cases with clear evidence of similar modes of action. If it is applied to mixtures with unknown modes of action, the user should be aware that this might result in an overestimation of the quality of confirmation by a factor of 2 or more. It should be also stressed that EC50-based TUs or TEQs only hold for this effect level. If applied for joint effect prediction (e.g. at a 20% effect level), an overestimation of confirmation quality by another factor of 2 may occur.
-
2)
Whenever possible, confirmation should be based on full dose–response relationships rather than on EC50 values. This allows prediction based on both IA and CA and extrapolation to different effect levels. Researchers who provide effect data on individual toxicants should publish full data sets rather than EC50 values.
-
3)
For some frequently applied toxicological endpoints, including genotoxicity and mutagenicity, the conceptual and mechanistic basis of mixture effect prediction is very weak. Thus, the present state of knowledge does not allow a quantitative estimate of contributions of individual compounds and of general confirmation quality.
Toxicity confirmation according to TIE
Effect confirmation in EDA has been discussed so far on the basis of chemical analytical and in vitro and in vivo effects, ignoring in situ exposure conditions and the relevance to organisms, populations and communities in situ. In order to confirm relevance under in situ or similar conditions, concepts derived from the toxicity identification evaluation (TIE) approach used by the US EPA [9] may be quite helpful and thus will be reviewed briefly. TIE was designed for application to effluents by the US EPA and provides a selection of well-defined confirmation procedures. The authors of the TIE guideline mention two major reasons why confirmation procedures are required. Firstly, during the characterisation and identification of toxicity, effluents are manipulated in a way that may “create artefacts that lead to erroneous conclusions about the cause of toxicity”. Thus, TIE confirmation must be based on the original sample and sample manipulations should be minimised or avoided as far as possible. Secondly, since the major point of concern for TIE is effluent control, confirmation should account for the variability of an effluent from sample to sample and from season to season. The following approaches are suggested:
-
The correlation approach is designed to show whether there is a consistent relationship between the concentration of the suspected toxicant(s) and the effluent toxicity. This concept assumes a sample with a spatial or time variance but an invariant major toxicant or a small set of toxicants that cause(s) the effect. Thus, if this major toxicant was correctly identified, reduced toxicity should be paralleled by reduced concentrations of this compound. This concept accounts for the specific task of the long-term control of time-variant effluents. It provides a helpful plausibility control for the identified toxicant and avoids sample manipulations that might impact upon matrix effects, including changes in bioavailability. It gives a semi-quantitative measure of how much of the toxicity can be explained by the toxicant by evaluating the correlation coefficient. Although designed for effluents, the correlation approach can provide a good basis for toxicity confirmation in sediments and surface waters as long as the set of responsible toxicants remains constant.
-
The symptom approach suggests that dissimilar symptoms observed in a biotest for the sample and the identified compound, respectively, exclude the identified compound as being a cause of the effect. Since many compounds might exhibit the same symptoms, the reverse clearly does not hold. Unfortunately, only a limited number of biotests allow the detection of different symptoms. Examples are in vivo chlorophyll a fluorescence [86], fish embryo testing [87] and behavioural toxicity syndromes in juvenile fathead minnows [88]. The symptom approach may provide a line of evidence for biological test systems that allow the discrimination of symptoms.
-
The species sensitivity approach suggests that the sensitivities of different species towards the sample and the toxicant of concern must be similar if TIE identified the correct compound. This approach assumes the application of in vivo testing of at least two species with different but comparable sensitivities. However, it might also be used for in vitro assays focusing on a similar endpoint, such as a battery of different test systems on mutagenicity and genotoxicity [89] or one test system with a battery of tester strains [90]. For species or test systems with very distinct sensitivities, we face the risk that both test systems detect different compounds, as shown for sediment extracts containing prometryn and methyl parathion. While green algae detected the herbicide prometryn, the insecticide methyl parathion was detected by daphnia [91].
-
The spiking approach is based on spiking samples with additional amounts of the toxicant of concern and retesting it. Doubling the concentration of the toxicant should halve the effect concentration of the sample. For aqueous samples this approach is almost generally applicable and integrates quantitative toxicity confirmation with proper consideration of matrix effects. A nice example of the successful application of this approach was the confirmation that PAHs cause the toxicity of motorway runoff to Daphnia pulex [52]. Possible applications of the spiking approach will be discussed below.
-
The mass balance approach ensures that the original toxicity of a sample is actually recovered in the fractions. This approach is applicable to situations in which toxicants can be removed from a sample and recovered in subsequent manipulation steps. This holds for example for aqueous samples where the toxicity can be removed with solid-phase extraction and subsequently fractionated and recovered by sequential elution. The resulting fractions are re-added to the previously extracted water sample individually, merged into a mixture and retested. Differences between the effect concentrations of the native sample and the reconstituted one indicate losses during the fractionation procedure. This cross-check is an excellent tool for the EDA of water samples. The basic principle can also be transferred to the EDA of the extracts; for instance, to account for losses during fractionation (e.g. by HPLC separations), mixtures based on the fractions should be reconstituted and retested for comparison with the original extract.
Hazard confirmation
The EDA of water, sediment or soil extracts based on in vivo toxicity in single-species tests or on sublethal in vitro effects may be regarded as a rather artificial system with great analytical power but with limited ecological relevance. Thus, it is challenging to confirm hazards resulting from key toxicants identified by EDA under realistic exposure conditions and for higher biological levels, such as whole organisms, populations and communities.
While toxicity confirmation in aqueous samples under realistic exposure conditions may be based on the TIE approaches discussed above, toxicity confirmation in solid samples such as soils and sediments is much more difficult. While compounds dissolved in water samples are generally regarded as being available to biota, bioavailability in soils and sediments is a major issue in hazard confirmation. As highlighted recently, the concept of bioavailability includes two complementary facets: (i) bioaccessibility, which discriminates molecules that readily desorb from sediments from a tightly bound pool of molecules that are not involved in partitioning with pore water or biota because of slow desorption kinetics; (ii) the equilibrium partitioning of accessible toxicants according to their chemical activity in different compartments [92].
Recently, new approaches for sediment TIEs under realistic exposure conditions have been suggested. They are based on sediment contact tests together with adsorbents that selectively remove bioaccessible fractions of specific groups of toxicants from pore water. Examples are zeolites for removing ammonia [93], ion-exchange resins for cationic metals [94] and for anionic arsenic and chromium [95], and powdered coconut charcoal for organic toxicants [96]. These are promising tools for toxicity characterisation, i.e. the discrimination of different types of toxicants. To be able to use them in toxicity identification and confirmation in the sense of EDA, two prerequisites have to be met. After loading, (i) the adsorbents need to be separated from the sediment, and (ii) the toxicants need to be recovered from the adsorbent for subsequent fractionation. Since EDA typically focuses on organic toxicants, these prerequisites must be shown to hold, particularly for charcoal. Unfortunately, charcoal cannot be separated from sediment and it does not allow sufficient recovery of adsorbed compounds. Thus, TENAX has been suggested as an alternative method of extracting bioaccessible sediment contaminants [97, 98]. The resin can be easily separated from sediments in suspension and extracted with organic solvents in order to subject the extracted mixture to EDA. This opens new possibilities for applying the spiking approach to toxicant confirmation to sediments. As presented recently, fractions and identified toxicants can be spiked back to the extracted sediments and retested under the same conditions as the native sediment [99]. Exposure in sediment contact tests is a result of the partitioning of bioaccessible fractions in the sediment–water–biota system, as driven by chemical activity. This approach is an elegant way to confirm sediment toxicants under realistic exposure conditions. It is typically applied to in vivo sediment contact tests. The confirmation of in vitro effects of sediment contaminants is possible on the basis of related in vivo effects and biomarkers. For example, the confirmation of exposure to potentially hazardous concentrations of estrogens via sediment using stimulation of embryo production in the freshwater mud snail Potaopyrgus antipodarum is a nice example of this approach [100]. In addition, a broad array of biochemical markers are available in invertebrates [101] as well as fish [102], which are closely linked to corresponding in vitro assays frequently used in EDA. Examples are DNA damage [103], vitellogenin in male fish as a biomarker for exposure to endocrine disruptors [104] and cytochrome P-450 activity in invertebrates [105] and fish [106].
The biomarkers, if determined in organisms in situ, can be correlated with concentrations of identified or expected key toxicants, providing additional lines of evidence according to the TIE correlation approach. A good example of that approach is the correlation of the contamination of sites in two tributaries of the River Llobregat (NE Spain) with nonylphenol and vitellogenin induction in fish [107]. Thus, dose-dependence of a biomarker induction provides a line of evidence for the confirmation of suggested key toxicants.
Recently, promising artificial substitutes for sediments (solid phases) in the spiking approach have been presented. These simulate the partitioning processes that occur in sediments in a standardised way and can generally be applied together with all in vivo and in vitro assays. These partition-based dosing techniques involve octadecylsilica disks [108], semi-permeable membrane devices (SPMDs) [109], polydimethylsiloxane (PDMS) films [110, 111] and coated stirring bars [112]. Partition-based dosing is believed to reflect the different bioavailabilities of the components of toxicant mixtures [109].
While bioavailability-directed extraction and dosing of sediment contaminants is always based on many assumptions and simulations that are hard to prove, direct EDA in tissues of benthic organisms per se only considers bioaccumulated toxicants. This approach has been applied for example to mussels, Mytilus edulis, from a field site contaminated with unresolved complex mixtures of aromatic hydrocarbons [113]. Tissue extracts and the fraction comprising aromatic hydrocarbons with 4–6 double-bond equivalents were confirmed to exhibit significant effects on the feeding rates of juvenile mussels of the same species. Another example is the identification of estrogenic compounds in deconjugated fish bile samples [114].
Concepts for confirming identified key toxicants at the community level are extremely rare. The most promising approach may be the concept of pollution-induced community tolerance (PICT) as a tool to confirm the impact of identified toxicants on, for example, periphyton [115] or plankton communities [116]. This concept is based on the fact that communities are often more tolerant to a compound they have been previously exposed to than communities without a history of exposure, and that individuals within a community that are sensitive to a specific toxicant will be replaced by more tolerant ones under toxicant exposure. This is believed to enhance the tolerance of the whole community [117, 118]. PICT can be quantified by a short-term metabolic test, such as photosynthesis inhibition.
One example of the applicability of this approach is a study conducted in the area of Bitterfeld (Spittelwasser). In sediment extracts, prometryn was identified as a key toxicant for algae [91]. In a subsequent PICT study, biofilms from the contaminated site and an uncontaminated (with respect to prometryn) site from the same area were tested for their sensitivity to prometryn (Fig. 3) [119]. The significantly increased tolerance of Spittelwasser biofilms confirmed the relevance of prometryn toxicity to the in situ community.

Comparison of concentration–response curves of prometryn derived with biofilms grown at river sites differing in environmental concentrations of the herbicide. Shifts in EC50 values indicate pollution-induced community tolerance (PICT). Modified after [117]
In conclusion, PICT provides the ability to confirm the hazards presented by individual toxicants identified by EDA at a community level. However, its application to confirmation is limited since it does not provide the ability to confirm effects on long-lived organisms with complex life cycles and it thus cannot link in vitro effects, which mostly focus on vertebrate toxicity, to community effects. Furthermore, no concept currently exists for using PICT for communities exposed to several toxicants, as is often found in the environment. A further limitation may be the development of co-tolerance to chemicals with similar modes of action, such as different uncouplers and photosynthetic inhibitors [117].
Conclusions
Confirmation is a crucial element in EDA. As a tiered approach it provides evidence for identified chemical structures and quantitative estimates of cause–effect relationships. Confirmation of all tiers is impeded by multiple restrictions, including small sample amounts, limited availability of neat standard chemicals, limited availability of spectra and retention data, a lack of information on concentration–effect relationships, a lack of clarity in joint effect prediction, and very limited tools for effect confirmation in situ. Thus, consistent guidance for the confirmation of toxicants at different levels of complexity is urgently needed. Important elements could be automated structure generation based on the analytical information gained during EDA, improved tools for structure elucidation and confirmation by LC–MS, QSARs and structure alerts for preselecting candidate toxicants, sound joint effect prediction based on full concentration–response relationships, novel approaches to including bioavailability in EDA confirmation, and advanced and more specific tools for effect confirmation in the field.
Abbreviations
- AhR:
-
arylhydrocarbon receptor
- AMDIS:
-
Automated Mass Spectral Deconvolution and Identification System
- BEQ:
-
benzo[a]pyrene equivalent quantity
- BP:
-
boiling point
- CA:
-
concentration addition
- CALUX:
-
chemical-activated luciferase expression
- ECX:
-
effect concentration required to achieve X% effect
- EDA:
-
effect-directed analysis
- EROD:
-
ethoxyresorufin-O-deethylase
- DNA:
-
deoxyribonucleic acid
- GC/MS:
-
gas chromatography with mass-selective detection
- IA:
-
independent action
- ICQ:
-
index of confirmation quality
- IEQ:
-
induction equivalent quantities
- IP:
-
identification points
- LC-Q-TOF-MS:
-
liquid chromatography with a hybrid quadrupole–time-of-flight mass spectrometer
- LSER:
-
linear solvation free-energy relationships
- NIST:
-
National Institute of Standards and Technology
- NMR:
-
nuclear magnetic resonance
- PAH:
-
polycyclic aromatic hydrocarbon
- PCB:
-
polychlorinated biphenyl
- PCDD/F:
-
polychlorinated dibenzo-p-dioxin and furan
- PDMS:
-
polydimethylsiloxane
- PICT:
-
pollution-induced community tolerance
- QSAR:
-
quantitative structure–activity relationship
- QSRR:
-
quantitative structure–retention relationship
- REP:
-
relative potency
- RI:
-
retention index
- RTL-W1:
-
rainbow trout liver cell line W1
- SPMD:
-
semipermeable membrane device
- TEQ:
-
toxicity equivalent quantity
- TIE:
-
toxicity identification evaluation
- TU:
-
toxic units
- US EPA:
-
United States Environmental Protection Agency
References
Schuetzle D, Lewtas J (1986) Anal Chem 58:1060A–1075A
Burgess RM (2000) Int J Environ Pollut 13:2–33
Brack W (2003) Anal Bioanal Chem 377:397–407
Hewitt LM, Marvin CH (2005) Mutat Res–Rev Mutat 589:208–232
Brack W, Klamer HJC, López de Alda MJ, Barceló D (2007) Environ Sci Pollut Res 14:30–38
Brack W, Schirmer K (2003) Environ Sci Technol 37:3062–3070
Norberg-King TJ, Mount DI, Durhan EJ, Ankley GT, Burkhard LP, Amato JR, Lukasewycz MT, Schubauer-Berigan MK, Anderson-Carnahan L (1991) Methods for aquatic toxicity identification evaluations. Phase I toxicity characterization procedures (EPA/600/6-91/003). United States Environmental Protection Agency, Washington, DC
Mount DI, Anderson-Carnahan L (1989) Methods for aquatic toxicity identification evaluations. Phase II toxicity identification procedures (EPA/600/3-88/035). United States Environmental Protection Agency, Washington, DC
Mount DI (1989) Methods for aquatic toxicity identification evaluation. Phase III toxicity confirmation procedures (EPA/600/3-88/036). United States Environmental Protection Agency, Washington, DC
Bailey HC, Elphick JR, Krassoi R, Lovell A (2001) Environ Toxicol Chem 20:2877–2882
Grote M, Brack W, Walter HA, Altenburger R (2005) Environ Toxicol Chem 24:1420–1427
Brack W, Bakker J, de Deckere E, Deerenberg C, van Gils J, Hein M, Jurajda P, Kooijman SALM, Lamoree MH, Lek S, López de Alda MJ, Marcomini A, Muñoz I, Rattei S, Segner H, Thomas K, von der Ohe PC, Westrich B, de Zwart D, Schmitt-Jansen M (2005) Environ Sci Pollut Res 12:252–256
Brack W, Kind T, Hollert H, Schrader S, Möder M (2003) J Chromatogr A 986:55–66
Meinert C, Moeder M, Brack W (2007) Chemosphere 70:215–223
Korytar P, Leonards PEG, de Boer J, Brinkman UAT (2005) J Chromatogr A 1086:29–44
Nukaya H, Yamashita J, Tsuji K, Terao Y, Ohe T, Sawanishi H, Katsuhara T, Kiyokawa K, Tezuka M, Oguri A, Sugimura T, Wakabayashi K (1997) Chem Res Toxicol 10:1061–1066
Oguri A, Shiozawa T, Terao Y, Nukaya H, Yamashita J, Ohe T, Sawanishi H, Katsuhara T, Sugimura T, Wakabayashi K (1998) Chem Res Toxicol 11:1195–1200
Shiozawa T, Tada A, Nukaya H, Watanabe T, Takahashi Y, Asanoma M, Ohe T, Sawanishi H, Katsuhara T, Sugimura T, Wakabayashi K, Terao Y (2000) Chem Res Toxicol 13:535–540
Belknap AM, Solomon KR, MacLatchy DL, Dube MG, Hewitt LM (2006) Environ Toxicol Chem 25:2322–2333
Christmann RF (1982) Environ Sci Technol 16:143A
National Institute of Standards and Technology (2007) Automated mass spectral deconvolution and identification system (AMDIS). NIST, Washington, DC (see http://chemdata.nist.gov/mass-spc/amdis/, last accessed 24 December 2007)
NIST/EPA/NIH (2005) Mass Spectral Library Version 2.0. US Department of Commerce, National Institute of Standards and Technology, Washington, DC
Benecke C, Grüner T, Kerber A, Laue R, Wieland T (1997) Fresenius’ J Anal Chem 359:23–32
Kerber A, Laue P, Meringer M, Rucker C (2005) Match–Commun Math Comp Chem 54:301–312
Kerber A, Laue R, Meringer M, Rücker C (2004) J Comput Chem Jpn 3:85–96
Kerber A, Laue R, Meringer M, Varmuza K (2001) Advances in mass spectrometry, vol 15. Wiley, New York
Eckel WP, Kind T (2003) Anal Chim Acta 494:235–243
Abraham MH (1993) J Phys Org Chem 6:660–684
Urbanczyk A, Staniewski J, Szymanowski J (2002) Anal Chim Acta 466:151–159
Vitha M, Carr PW (2006) J Chromatogr A 1126:143–194
Platts JA, Butina D, Abraham MH, Hersey A (1999) J Chem Inf Comp Sci 39:835–845
Schüürmann G, Ebert RU, Kuehne R (2006) Chimia 60:691–698
Brack W, Schirmer K, Erdinger L, Hollert H (2005) Environ Toxicol Chem 24:2445–2458
Petrovic M, Barcelo D (2006) J Mass Spectrom 41:1259–1267
Bobeldijk I, Vissers JPC, Kearney G, Major H, van Leerdam JA (2001) J Chromatogr A 929:63–74
Grung M, Lichtenthaler R, Ahel M, Tollefsen KE, Langford K, Thomas KV (2007) Chemosphere 67:108–120
EC (2002) Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and interpretation of results (2002/657/EC). European Commission, Brussels
Pérez S, Eichhorn P, Barceló D (2007) Anal Chem (in press)
Picó Y, la Farré M, Soler C, Barceló D (2007) Anal Chem (in press)
Farre M, Kuster M, Brix R, Rubio F, Alda MJL, Barcelo D (2007) J Chromatogr A 1160:166–175
Picó Y, la Farré M, Soler C, Barceló D (2007) J Chromatogr A (in press)
Braga RS, Barone PMVB, Galvao DS (1999) J Mol Struct–Theochem 464:257–266
Liu HX, Papa E, Gramatica P (2006) Chem Res Toxicol 19:1540–1548
von der Ohe PC, Kuhne R, Ebert RU, Altenburger R, Liess M, Schuurmann G (2005) Chem Res Toxicol 18:536–555
Fang H, Tong WD, Branham WS, Moland CL, Dial SL, Hong HX, Xie Q, Perkins R, Owens W, Sheehan DM (2003) Chem Res Toxicol 16:1338–1358
Arulmozhiraja S, Morita M (2004) Chem Res Toxicol 17:348–356
Estrada E, Molina E (2006) J Mol Graph Model 25:275–288
Enslein K, Gombar VK, Blake BW (1994) Mut Res 305:47–61
Altenburger R, Nendza M, Schuurmann G (2003) Environ Toxicol Chem 22:1900–1915
Faust M, Altenburger R, Backhaus T, Bodeker W, Scholze M, Grimme LH (2000) J Environ Qual 29:1063–1068
Swartz RC, Schults DW, Ozretich RJ, Lamberson JO, Cole FA, DeWitt TH, Redmond MS, Ferraro SP (1995) Environ Toxicol Chem 14:1977–1987
Boxall ABA, Maltby L (1997) Arch Environ Contam Toxicol 33:9–16
Clemons JH, Dixon DG, Bols NC (1997) Chemosphere 34:1105–1119
Jung KJ, Klaus T, Fent K (2001) Environ Toxicol Chem 20:149–159
Tillitt DE, Giesy JP, Ankley GT (1991) Environ Sci Technol 25:87–92
Willett KL, Gardinali PR, Sericano JL, Wade TL, Safe SH (1997) Arch Environ Contam Toxicol 32:442–448
Brown DJ, Chu M, Overmeire IV, Chu A, Clark GC (2001) Organohal Comp 53:211–214
Machala M, Vondracek J, Blaha L, Ciganek M, Neca J (2001) Mutat Res 497:49–62
van den Berg M, Birnbaum L, Bosveld ATC, Brunstrom B, Cook P, Feeley M, Giesy JP, Hanberg A, Hasegawa R, Kennedy SW, Kubiak T, Larsen JC, van Leeuwen FXR, Liem AKD, Nolt C, Peterson RE, Poellinger L, Safe S, Schrenk D, Tillitt D, Tysklind M, Younes M, Waern F, Zacharewski T (1998) Environ Health Persp 106:775–792
Vondracek J, Kozubik A, Machala M (2002) Toxicol Sci 70:193–201
Houtman CJ, Van Houten YK, Leonards PG, Brouwer A, Lamoree MH, Legler J (2006) Environ Sci Technol 40:2455–2461
Blaha L, Kapplova P, Vondracek J, Upham B, Machala M (2002) Toxicol Sci 65:43–51
Madill REA, Brownlee BG, Josephy PD, Bunce NJ (1999) Environ Sci Technol 33:2510–2516
Durant JL, Busby WF, Lafleur AL, Penman BW, Crespi CL (1996) Mutat Res 371:123–157
Altenburger R, Walter H, Grote M (2004) Environ Sci Technol 38:6353–6362
Brack W, Segner H, Möder M, Schüürmann G (2000) Environ Toxicol Chem 19:2493–2501
Brack W, Schirmer K, Kind T, Schrader S, Schüürmann G (2002) Environ Toxicol Chem 21:2654–2662
Payne J, Rajapakse N, Wilkins M, Kortenkamp A (2000) Environ Health Persp 108:983–987
Faust M, Altenburger R, Backhaus T, Blanck H, Boedeker W, Gramatica P, Hamer V, Scholze M, Vighi M, Grimme LH (2003) Aquat Toxicol 63:43–63
Walter H, Consolaro F, Gramatica P, Scholze M, Altenburger R (2002) Ecotoxicology 11:299–310
Tammer B, Lehmann I, Nieber K, Altenburger R (2007) Toxicol Lett 170:124–133
Kortenkamp A, Altenburger R (1999) Sci Total Environ 233:131–140
Hilscherova K, Kannan K, Kang YS, Holoubek I, Machala M, Masunaga S, Nakanishi J, Giesy JP (2001) Environ Toxicol Chem 20:2768–2777
Gale RW, Long ER, Schwartz TR, Tillitt DE (2000) Environ Toxicol Chem 19:1348–1359
Putzrath RM (1997) Regul Toxicol Pharmacol 25:68–78
Villeneuve DL, Blankenship AL, Giesy JP (2000) Environ Toxicol Chem 19:2835–2843
Rosenkranz HS, McCoy EC, Sanders DR, Butler M, Kiriazides DK, Mermelstein R (1980) Science 209:1039–1042
Møller M, Alfheim I, Larssen S, Mikalsen A (1982) Environ Sci Technol 16:221–225
Fernandez P, Grifoll M, Solanas AM, Bayona JM, Albaiges J (1992) Environ Sci Technol 26:817–829
Durant JL, Lafleur AL, Plummer EF, Taghizadeh K, Busby WF, Thilly WG (1998) Environ Sci Technol 32:1894–1906
Erdinger L, Dorr I, Durr M, Hopker KA (2004) Mutat Res–Genet Toxicol Environ Mutagen 564:149–157
Marvin CH, Tessaro M, McCarry BE, Bryant DW (1994) Sci Tot Environ 156:119–131
White PA (2002) Mut Res 515:85–98
Haugen DA, Peak MJ (1983) Mut Res 116:257–269
Zeiger E, Pagano DA (1984) Environ Mutagen 6:683–694
Brack W, Frank H (1998) Ecotox Environ Saf 40:34–41
Ensenbach U (1998) Fres Environ Bull 7:531–538
Drummond RA, Russom CL (1990) Environ Toxicol Chem 9:37–46
Pellacani C, Buschini A, Furlini M, Poli P, Rossi C (2006) Aquat Toxicol 77:1–10
Nikoyan A, De Meo M, Sari-Minodier I, Chaspoul F, Gallice P, Botta A (2007) Mutat Res–Genet Toxicol Environ Mutagen 626:88–101
Brack W, Altenburger R, Ensenbach U, Möder M, Segner H, Schüürmann G (1999) Arch Environ Contam Toxicol 37:164–174
Reichenberg F, Mayer P (2006) Environ Toxicol Chem 25:1239–1245
Burgess RM, Perron MM, Cantwell M, Ho KT, Serbst JR, Pelletier E (2004) Arch Environ Contam Toxicol 47:440–447
Burgess RM, Cantwell MG, Pelletier MC, Ho KT, Serbst JR, Cook HF, Kuhn A (2000) Environ Toxicol Chem 19:982–991
Burgess RM, Perron MM, Cantwell MG, Ho KT, Pelletier MC, Serbst JR, Ryba SA (2007) Environ Toxicol Chem 26:61–67
Ho KT, Burgess RM, Pelletier MC, Serbst JR, Cook H, Cantwell MG, Ryba SA, Perron MM, Lebo J, Huckins JN, Petty J (2004) Environ Toxicol Chem 23:2124–2131
Cornelissen G, Rigterink H, ten Hulscher DEM, Vrind BA, van Noort PCM (2001) Environ Toxicol Chem 20:706–711
Schwab K, Brack W (2007) J Soil Sediments 7:178–186
van den Heuvel-Greve MJ, Kooman H, Hermans J, Bakker J (2007) A TIE pilot study using in vivo bioassay with the estuarine amphipod, Corophium volutator, as a first approach to in vivo EDA. Poster presentation at 17th Annual Meeting of SETAC Europe, Porto, Portugal, 20–24 May 2007
Duft M, Schulte-Oehlmann U, Weltje L, Tillmann M, Oehlmann J (2003) Aquat Toxicol 64:437–449
Hyne RV, Maher WA (2003) Ecotox Environ Saf 54:366–374
Hinton DE, Kullman SW, Hardman RC, Volz DC, Chen PJ, Carney M, Bencic DC (2005) Mar Pollut Bull 51:635–648
Shugart LR (2000) Ecotoxicology 9:329–340
Hutchinson TH, Ankley GT, Segner H, Tyler CR (2006) Environ Health Persp 114:106–114
Fisher T, Crane M, Callaghan A (2003) Ecotox Environ Saf 54:1–6
Machala M, Dušek L, Hilscherová K, Kubínová R, Jurajda P, Neca J, Ulrich R, Gelnar M, Studnicková Z, Holoubek I (2001) Environ Toxicol Chem 20:1141–1148
Solé M, López de Alda MJ, Castillo M, Porte C, Ladegaard-Pedersen K, Barceló D (2000) Environ Sci Technol 34:5076–5083
Mayer P, Wernsing J, Tolls J, de Maagd PGJ, Sijm DTHM (1999) Environ Sci Technol 33:2284–2290
Heinis LJ, Highland TL, Mount DR (2004) Environ Sci Technol 38:6256–6262
Brown RS, Akhtar P, Akerman J, Hampel L, Kozin IS, Villerius LA, Klamer HJC (2001) Environ Sci Technol 35:4097–4102
Kiparissis Y, Akhtar P, Hodson P, Brown RS (2003) Environ Sci Technol 37:2262–2266
Bandow N, Altenburger R, Paschke A, Brack W (2007) PDMS coated stirring bars—a new method to include the bioavailability in the effect-directed analysis of contaminated sediments. Presentation at 17th Annual Meeting of SEATC Europe, Porto, Portugal, 20–24 May 2007
Donkin P, Smith EL, Rowland SJ (2003) Environ Sci Technol 37:4825–4830
Houtman CJ, Van Oostven AM, Brouwer A, Lamoree MH, Legler J (2004) Environ Sci Technol 38:6415–6423
Schmitt-Jansen M, Altenburger R (2005) Environ Toxicol Chem 24:304–312
Petersen S, Gustavson K (1998) Aquat Toxicol 40:253–264
Blanck H (2002) Human Ecol Risk Assess 8:1003–1034
Schmitt-Jansen M, Veit U, Dudel G, Altenburger R (2007) Basic Appl Ecol DOI 10.1016/j.baae.2007.08.008
Schmitt-Jansen M, Reiners S, Altenburger R (2006) UWSF-Z Umweltchem Ökotoxikol 16:85–91
Acknowledgements
This study was funded by the European Union in the framework of the Integrated Project MODELKEY (Contract 511237-GOCE).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brack, W., Schmitt-Jansen, M., Machala, M. et al. How to confirm identified toxicants in effect-directed analysis. Anal Bioanal Chem 390, 1959–1973 (2008). https://doi.org/10.1007/s00216-007-1808-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-007-1808-8