
Abstract
The weakest step in the analytical procedure for speciation analysis is extraction from a biological material into an aqueous solution which undergoes HPLC separation and then simultaneous online detection by elemental and molecular mass spectrometry (ICP–MS/ES-MS). This paper describes a study to determine the speciation of arsenic and, in particular, the arsenite phytochelatin complexes in the root from an ornamental garden plant Thunbergia alata exposed to 1 mg As L−1 as arsenate. The approach of formic acid extraction followed by HPLC–ES-MS/ICP–MS identified different AsIII–PC complexes in the extract of this plant and made their quantification via sulfur (m/z 32) and arsenic (m/z 75) possible. Although sulfur sensitivity could be significantly increased when xenon was used as collision gas in ICP–qMS, or when HR-ICP–MS was used in medium resolution, the As:S ratio gave misleading results in the identification of AsIII–PC complexes due to the relatively low resolution of the chromatography system in relation to the variety of As–peptides in plants. Hence only the parallel use of ES-MS/ICP–MS was able to prove the occurrence of such arsenite phytochelatin complexes. Between 55 and 64% of the arsenic was bound to the sulfur of peptides mainly as AsIII(PC2)2, AsIII(PC3) and AsIII(PC4). XANES (X-ray absorption near-edge spectroscopy) measurement, using the freshly exposed plant root directly, confirmed that most of the arsenic is trivalent and binds to S of peptides (53% As–S) while 38% occurred as arsenite and only 9% unchanged as arsenate. EXAFS data confirmed that As–S and As–O bonds occur in the plants. This study confirms, for the first time, that As–peptides can be extracted by formic acid and chromatographically separated on a reversed-phase column without significant decomposition or de-novo synthesis during the extraction step.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Knowledge of arsenic speciation in biota has increased enormously over the last two decades due to advances in analytical chemistry, particularly in the area of element-specific techniques such as ICP–MS. At the end of the 20th century only arsenite, arsenate, and some organoarsenicals such as arsenobetaine, arsenosugars or the methylated species were known to occur in biological samples [1]. It is only recently that the interaction between arsenic and sulfur was detected in low-molecular-mass thio-organoarsenical compounds [2–5] and in the form of arseno-peptides [6, 7] such as AsIII-phytochelatins (AsIII–PCs). Phytochelatins (PCs) are cysteine-containing peptides formed by terrestrial plants to cope with toxic amounts of essential and non-essential elements such as Cu, Zn, As, Cd, or Hg ions. The general structure of PCs can be described by the following formula (γ-Glu–Cys) n –Gly (n = 2–11), whereas glycine (Gly) can be replaced by glutamic acid (Glu), β-alanine (Ala), serine (Ser), or a hydroxyl group to form the iso-PCs. PCs seem to play an important role in the detoxification and homeostasis processes of plants, since metal ions complexed via the sulfhydryl groups of their cysteine residues are less toxic than the free ions [8–12].
The first demonstration that arsenite forms complexes with PC in vivo utilized a hyphenated analytical technique in which reversed-phase HPLC was simultaneously coupled to ICP–MS and ES-MS [13]. The use of HPLC provides the separation of the different species found in plant tissues whereas the element-specific information is given by ICP–MS. To get a full set of data about the organo-arsenic speciesFootnote 1 it is necessary to use a detector that provides molecular specific information, for example ES-MS [14, 26]. An important point in speciation is the choice of methods for extraction and analysis, since they must not change the species of interest. Arsenite phytochelatins are known to be unstable during sample preparation and chromatography. Degradation of compounds during speciation analysis not only leads to erroneous results but also to in-vitro de-novo formation during extraction. The latter can occur as a result of mixing different cell compartments and/or the creation of completely different conditions in the extract.
In the past the speciation of phytochelatins was limited to the quantification of their complexed arsenite by HPLC–ICP–MS and their identification using ES-MS. Nothing could be said about the concentrations of free reduced and oxidised PCs in plant extracts. For their quantification using ICP–MS the only suitable heteroatom in the peptide chains is sulfur (m/z = 31.9721) which, however, exhibits strong \( O^{ + }_{2} \) (m/z = 31.9898) interferences. There are different approaches to quantify sulfur-containing compounds, including the use of high resolution ICP–MS, the removal of the solvent by a desolvation system, or the use of reaction/collision cells pressurised with either a reaction gas such as oxygen or relatively “heavy” collision gases such as Ar or Xe. But still the result is only as good as the weakest link in the speciation analysis. These advanced techniques can only give insight into the speciation of the extract but not into the speciation in the plant cells. Therefore other analytical techniques which do not require an extraction step, for example XANES (X-ray absorption near-edge spectroscopy) and EXAFS (extended X-ray absorption fine structure) are necessary. The use of XAS (X-ray absorption spectroscopy) methods gives information about the oxidation state of arsenic and its direct chemical environment in the plant cells.
The aim of this paper is to show which information, with regard to arsenic speciation in plants, can be obtained by use of different analytical techniques of increasing sophistication, using as an example an arsenic-exposed ornamental garden plant Thunbergia alata commonly known as “Black Eyed Susan”.
Materials and methods
Plant exposure experiment
Thunbergia alata was grown from commercial seeds. The plants were grown in vermiculite at 24°C in a greenhouse and fertilized weekly using commercial full-fertilizer with low background As concentration (<0.1 ng mL−1) for more than one month. One week before exposure to As fertilisation was stopped. More than twenty individual plants were exposed to 1 mg arsenic L−1 AsV for 24 h at slightly different times.
Chemicals
All chemicals used were of analytical grade or better. Ultra-pure water was used throughout (Elga UK). Formic acid (100%) was from Fisher, methanol (HPLC-grade) from Rathburn (UK), sodium-dimethylarsinic acid (98%, DMAV), used as calibration standard for quantification, and sodium arsenite (AsIII) were obtained from ChemService (USA). Nitric acid (65%) and hydrogen peroxide (32%) used in total As analysis were obtained from Fluka (UK) and elemental standards (arsenic and rhodium as internal standard) were from High Purity Standards (Charleston, USA). Disodiumhydrogenarsenate heptahydrate (AsV) and sodiumdihydrogenorthophosphate dihydrate were supplied from BDH. l-Methionine was obtained from Sigma, and PC2 and PC3 from Clonestar Biotech (Czech Republic).
Standards: AsIII–PC2 and AsIII–PC3 mixture
The free reduced phytochelatins were dissolved in deionised water with sulfur concentrations of approximately 900 nmol g−1 for PC2 and 700 nmol g−1 for PC3. For the synthesis of AsIII–PCs an AsIII solution containing approx. 2.2 μmol As g−1 was prepared in deionised water using NaAsO2. The formation of the AsIII–PC complexes was achieved by adding 0.9 g As(III) solution to 0.1 g of the PC stock solutions. The preparation of these standards was performed under nitrogen atmosphere to avoid oxidation of the PCs. Structures of these complexes are indicated in Fig. 1.

Structures of arsenite phytochelatins complexes. Two isoforms exist for AsIII(PC2)2 while four exist for AsIIIPC4. GLU, (glutamic acid); CYS, (cysteine); GLY, (glycine)
Extraction methods
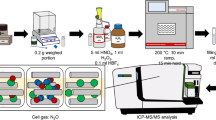
A detailed description of the sample preparation for speciation and total As analysis, and detailed analytical methods, can be found in Raab et al. [26]. Briefly, fresh plant roots were ground under liquid nitrogen after removal of external As by use of phosphate-containing solution [15]. A sub-sample was extracted with 1% formic acid for 60 min at 1°C and filtered before analysis of AsIII–PCs. Analysis was performed within 4 h of extraction. Because of the procedural obstacle that a fresh extract had to be measured, it was not possible to use the same extract for all analytical methods. Hence, different plants of similar age were always extracted prior to the analysis but after the 24 h exposure to arsenate.
Analytical methods
HPLC-ICP–qMS/ES-ion-trap MS
For separation of the As species present in plant extracts a reversed-phase column (C18, Ace 5 C18, Advanced Chromatography Technologies, Aberdeen, UK) was used. A gradient of 0.5% formic acid in water and 0.5% formic acid in methanol was used for the separation and the eluent-flow was split after the column (one part into the ICP–MS (Agilent 7500c, Agilent USA) and eight parts into the ES-trapMS (MSD XCT, Agilent USA)). The ES-trapMS was used in positive ultrascan mode and MS2 spectra were recorded when signal intensities were higher than 50.000 counts. The ICP–MS was used in normal mode using platinum cones and 6% oxygen with a microconcentric nebulizer, and 100 μg L−1 rhodium solution was added before the nebulizer as continuous internal standard (Table 1). Arsenic on m/z 75, (m/z 77 for chloride interference), sulfur on m/z 34, and rhodium (m/z 103) were monitored. All quantification was done using the ICP–MS signal on m/z 75; for speciation DMAV (peak areas vs. concentration) was used as calibration species.
HPLC–HR-ICP–MS and HPLC–Orbitrap MS
For separation of the As species present in plant extracts a reversed-phase column (C18, Ace 5 C18, Advanced Chromatography Technologies (Aberdeen, UK) for Orbitrap / Element 2 experiments) was used. A gradient of 0.5% formic acid in water and 0.5% formic acid in methanol was used for the separation. The column was coupled either to the ICP–MS or to the ES-MS. A high-mass-resolution ICP–MS (Element 2, ThermoFisher) was used with platinum cones, Meinhard nebulizer and oxygen and optimised as necessary (Table 1). It was used in medium-resolution mode for the measurement of sulfur (m/z 32) and arsenic (m/z 75). All quantification was done using the ICP–MS signal on m/z 75 and m/z 32, for speciation DMAV and sulfate (peak areas vs. concentration) were used as calibration species.
The Orbitrap instrument was used in positive mode with an ES source in high-resolution mode with external calibration (mass range 100 to 1500 m/z). No parallel use of the detectors was possible, therefore the sample was injected twice, first on the HPLC–HR-ICP–MS and as soon as the measurement was finished on the HPLC–ES-Orbitrap-MS.
HPLC–(ICP–qMS [with xenon] / ES-MS)
For separation of the As species present in the AsIII–PC standard solutions and plant extracts a reversed-phase column (Eclipse XDB-C18, 4.6 × 150 mm, Agilent) was used. Throughout all experiments the HPLC was coupled to an ES-MS (Agilent 1100, MSD, Agilent, USA) and an Agilent 7500c quadrupole ICP–MS with an octupole collision cell system (Agilent, USA). In order to remove the \( O^{ + }_{2} \) interferences the collision cell was pressurised with xenon. The eluent-flow was split 1:1 after the column (Table 1). All quantification was done using the ICP–MS signal on m/z 75 and m/z 32; for speciation DMAV and methionine (peak areas vs. concentration) were used as calibration species.
HPLC–(ICP–qMS [with oxygen] / ES-MS)
Throughout all experiments the HPLC, using a reversed-phase column (Eclipse XDB-C18, 4.6 × 150 mm, Agilent) was coupled to an ES-MS (Agilent 1100, MSD, Agilent, USA) and an Agilent 7500c quadrupole ICP–MS with an octupole-collision cell system (Agilent, USA). The collision cell was pressurised with oxygen which allows the quantification of sulfur on m/z 48 (SO+). For the determination of the detection limits of sulfur as SO+ and arsenic on m/z 48, 75 (As) and 91 (AsO+) methionine and DMAV were used. The standards were made up at a concentration of approximately 500 μg As and S L−1 and measured three times at optimised ICP–qMS settings (Table 1).
XANES/EXAFS
X-ray absorption spectra at the As K-edge were collected on Station 16.5 at the CLRC (now STFC) Daresbury SRS operating at 2 GeV with an average current of 150 mA, using a vertically focusing mirror and a sagitally bent focusing Si(220) double-crystal monochromator detuned to 70% transmission to minimise harmonic contamination. For the four model compounds data were collected in transmission mode at ambient temperature. Data for the other samples were collected with the station operating in fluorescence mode using an Ortec 30 element solid-state Ge detector. Experiments were performed in a liquid nitrogen-cooled cryostat. Several scans were collected and summed for the each sample. XANES (X-ray absorption near edge structure) data were fitted using a linear combination of XANES spectra collected from model systems. The models used were sodium arsenate (AsV coordinated by four oxygen atoms at 1.68 Å), sodium arsenite (AsIII coordinated by three oxygen atoms at 1.79 Å), dimethyl arsenic acid, DMAV (As coordinated by two oxygen atoms at 1.62 Å and two carbon atoms at 1.91 Å), and a sodium glutathione complex (AsIII coordinated by three sulfur atoms at ca. 2.26 Å), which were taken from crystallographic sources [16–19].
Results and discussion
Identification using HPLC-ICP–qMS/ES-MS
A reversed-phase HPLC method was optimized to separate the small unbound peptides from their arsenic complexes, found in the extracts of T. alata roots (Fig. 2). The separation by RP chromatography showed the formation of AsIII–PC complexes using ES-MS detection in positive-ion monitoring mode (Fig. 2). Simultaneous use of low resolution ICP–qMS in normal mode enabled the detection and quantification of arsenic on m/z 75, but the sulfur detection limit on 34-S was too high to detect any sulfur compounds in the standard solutions or the root extracts (except free sulfate and glutathione) (data not shown). The detection limit for As (m/z 75) in the configuration used for these experiments was approximately 3 μg As L−1, the detection limit for S (m/z 34) was estimated to be approximately 100 mg L−1. This method has been successfully used for identification and quantification of the AsIII–PC in plant extracts [6, 13, 26, 27]. However, when ICP–qMS is used solely as a detector the identification of the AsIII–PCs or free PCs via their sulfur is not possible in physiologically relevant concentrations due to its high detection limits for sulfur. These high detection limits for sulfur are caused by its low ionisation efficiency, and the high background contributed by ubiquitous oxygen (present as \( O^{ + }_{2} \) on m/z 32 and 34) had to be overcome in order to enable the identification of As–S peptides and the quantification of arsenite-bound and unbound, reduced and oxidised PCs in standards and plant extracts.

Use of RP-HPLC coupled simultaneously to ES-MS and ICP–qMS for determination of arsenite phytochelatins complexes in root extracts from Thunbergia alata exposed to arsenate. ICP–MS is used as indicator of arsenic-containing peptides and for quantification (m/z 75), while ES-MS signals indicate the different AsIII–PC complexes according to the protonated molecular masses
Three different approaches were used to overcome the polyatomic interferences: The first strategy, in which the aerosol is dried, has not been studied in depth. Initial results however showed that membrane desolvation leads to element species-specific loss during the drying process. Since species-specific behaviour in the sample introduction system is counteracting the use of ICP–MS to quantify element species without molecular-identical standards, this approach was not further pursued.
The second approach was the use of collision-induced dissociation and charge transfer of \( O^{ + }_{2} \) by pressurising the collision-cell of the Agilent 7500c with xenon. The third approach used a high-resolution (HR)-ICP–MS with the ability to separate the signals derived from 32S+ (m/z 31.9721) and \( O^{ + }_{2} \) (m/z 31.9898). The last two approaches led to much reduced detection limits for sulfur (Table 1). First experiments to improve the detection limits for sulfur by pressurising the collision cell of the Agilent 7500c system with oxygen and thereby move the detection mass from m/z 32 to 48 (SO+) resulted in no further improvement (Table 1), because the applied kinetic energy discrimination (KED) not only prevented the newly formed interferences, e.g., \( O^{ + }_{3} \), from reaching the detector, but also SO+. The use of oxygen as reaction gas did not improve the detection limit of arsenic on either m/z 75 or 91.
Identification using HPLC–ICP–qMS/ES-MS with xenon as collision gas
The use of xenon as collision gas removed the \( O^{ + }_{2} \) interferences on m/z 32 which made the quantification of sulfur-containing compounds using HPLC–ICP–qMS possible, but also affected the intensity of analyte ions by collisional and charge-transfer processes [20–24], which led to a general decrease in signal intensities. In Table 1 the instrument parameters for ICP–qMS (xenon) and the limits of detection for the four different type of instrumentation are given. They were calculated from three times the standard deviation of the noise level under consideration of the element response during the chromatographic runs using DMAV and methionine or sulfate as standard species.
AsIII–PC2 and AsIII–PC3 standards (approx. 2 and 5 μg complex g−1) were used to test the suitability of the HPLC–ICP–qMS (xenon) setup for simultaneous detection of As (m/z 75) and S (m/z 32) with simultaneous detection of the eluting compounds by ES-MS. The HPLC–ICP–qMS spectra showed signals for AsIII–PC2 at 7 and 8 min, for AsIII–PC3 at 12.4 min, for inorganic As at 1.5 min, and for co-eluting As and S species (AsIII–PC2 at 4.5 min and AsIII–PC3 at 11.4 and 13.7 min). In addition, unbound sulfur-containing peptides such as reduced and oxidized PC2 or PC3 could be identified (Fig. 3).

Chromatograms of: (a) AsIII–PC2 mixture and (b) AsIII–PC3 mixture, obtained by use of HPLC dual MS (ICP–qMS [xenon]/ES-MS). (a) peak 1 indicate the occurrence of AsIII(OH)PC2 with protonated molecular mass m/z 630, while free PC2 (red., m/z 540 and (ox) m/z 538) elute later. (b) peak at 12 min is unidentified, while free-PC3 (m/z 772, and m/z 770) elute slightly earlier than the AsIII–PC3 complex indicated by the m/z 844 signal
For identification of the arsenic sulfur complexes, it has been suggested that the As:S ratio could be a reliable indicator for their identification [25]. In the mixture of AsIII and PC2 the formation of AsIII–(PC2)2 with a As:S ratio of 1:4 was expected. The formation of this complex was shown to occur using a mixture of AsIII, PC2, GSH and PC3 [26]. The ratio determined from the chromatogram was, however, only 1:2.1 (3.74 nmol As g−1; 7.18 nmol S g−1), implying that only one PC2 molecule was involved in the complexation of arsenite in the prepared standard. This assumption was supported by the ES-MS chromatogram which showed a signal at m/z 630 co-eluting with the As and S peaks, which would be the protonated molecular mass of AsIII(OH)PC2. The final proof of its presence was obtained from its MS2 spectrum, which showed signals at m/z 612 (loss of water), 537 (loss of water and arsenic), and 483 (loss of water and glutamic acid) (data not shown). The detection limit for sulfur using xenon as collision gas was sufficient to detect unbound cysteine-containing peptides—free reduced PC2 (m/z 540, RT: 8 min) and oxidised PC2 (m/z 538, RT: 7 min).
The AsIII–PC3 standard (expected As:S ratio 1:3) contained next to the inorganic arsenic two other arsenic species (Fig. 3). For the one eluting at 11.4 min no information about the As:S ratio was available since the sulfur signal was too small for proper integration. An As:S ratio of 1:3.1 (6.13 nmol As g−1; 18.85 nmol S g−1) gave reason to suppose that the third arsenic peak at 13.7 min was caused by AsIII–PC3. At the same retention time the ES-MS gave a signal on m/z 844 confirming the eluting AsIII–PC3 complex. Here again the m/z 32 trace for sulfur showed the evidence of unbound cysteine-containing peptides such as reduced PC3 (m/z 772, RT: 12.4 min) and oxidised PC3 (m/z 770, RT: 12.4 min).
The root extract of AsV-exposed T. alata, which was previously been shown to produce large amounts of AsIII–PC complexes, was run under the same conditions as the AsIII–PC standards. The ICP–qMS spectra showed a co-eluting peak of arsenic and sulfur at 13.8 min (Fig. 4). The retention time gave reason to suggest the occurrence of AsIII–PC3 since the signal showed almost the same retention time as the corresponding standard. But the found As:S ratio of 1:2.4 (±0.2; four replicates) did not fit with the theoretical expected one of 1:3. The simultaneous use of ES-MS confirmed unequivocally the presence of AsIII–PC3 since its protonated molecular mass (m/z 844) was co-eluting with the arsenic and sulfur signals at 13.8 min. Hence the accuracy of the As:S ratio was questioned. One reason for this ambiguity might have been a matrix effect which resulted in an unsteady baseline and made the integration more difficult even though the complex was present (in the extract) in relatively high concentrations (4 nmol As g−1; 10 nmol S g−1). The other reason might be the co-elution of other arsenic species. The number of possible phytochelatin complexes can be enormous, since not only the standard PCs as seen in Fig. 1 can be produced by plants but also iso-PCs and EC-PCs with alternating amino acids replacing glycine. Therefore low chromatographic resolution could have been responsible for incorrect As:S ratios. This means that even if AsIII–PC3 standards are available, the correct identification can not easily be done by measuring the As:S ratios at certain retention times using ICP–qMS.

Arsenic and sulfur speciation in the root extract of Thunbergia alata, after exposure for 24 h to a 1 ppm As(V) solution, using ICP–qMS (xenon). Free-PC2 is barely detected at m/z 32, while the main complex AsIII–PC3 is clearly identified by m/z 75 and 32 in the ICP–MS and m/z 844 in the ES-MS
Identification using HPLC–HR-ICP–MS/ES-Orbitrap MS
Measurement of T. alata root extract using HR-ICP–MS showed the presence of at least 24 S-containing species, of which only cysteine, glutathione, and some free PCs can be assigned (Fig. 5). Additionally, at least seven As species were determined by HR-ICP–MS, which were not all baseline separated, which makes the use of As:S ratio difficult. For example the integration of the As and S-signals at 20.98 and 21.24 min was hampered by the low chromatographic resolution. ES-MS data from the ES-Orbitrap showed that these signals correspond to oxidised PC3 (m/z 770) and the AsIII–PC3 complex (m/z 844). The measured As:S ratio for the AsIII–PC3 was 1:3.1, similar to that found for the separation of the AsIII–PC3 standard using ICP–qMS (xenon). Hence, the enhanced detection limits for sulfur made it possible to determine correctly the occurrence of AsIII–PC3 in the root extract. The extract also contained one of the AsIII-(PC2)2 isoforms eluting at 23.5 min (measured As:S ratio 1:3.6) and AsIII–PC4 (measured As:S ratio 1:3.7). In both cases a ratio of 1:4 was expected. The difference between expected and measured ratio can have different reasons. For one there could be another unresolved As-S compound with a smaller As:S ratio co-eluting, for peak-area integration is somewhat difficult due to the low resolution. Although the HR-ICP–MS has the detection limit for S needed for real samples and could therefore identify the correct As:S ratio for the most abundant arsenic phytochelatin complex (AsIII–PC3), it was unable to predict the stoichiometry for the less abundant arseno complexes. Improvements of the chromatographic method would now be necessary to resolve to baseline all the different S-containing compounds. Finally, it could be concluded that the applicability of using the As:S ratio as an indication for the arsenic peptide when only ICP–MS is used as the detector is rather limited in real samples and should be used carefully for As–S complexes.

HPLC–HR-ICP–MS/ES-Orbitrap MS of a freshly prepared formic acid extract from a root of T. alata exposed to 1 mg As L−1, as arsenate, 24 h. (a) shows the traces of arsenic (m/z 75) and sulfur (m/z 32). 1, arsenate, arsenite, and sulfate; 2, cysteine; 3, glutathione; 4, free-PC3; 5, AsIII–PC3; 6, AsIII–PC4 (four isomers). (b) ES-MS of AsIII–PC3, accurate mass main peaks (844.0917, error −1.3 ppm of theoretical mass; 845.0947, error −1.65 ppm). (c) MS2 of m/z 844 → m/z 715 (loss of glutamic acid). (d) ES-MS of AsIII–PC4, accurate mass of two main peaks 1076.1437, error −0.74 ppm; 1077.1467, error −1.11 ppm). (e) MS2 of m/z 1076 → m/z 947 (loss of glutamic acid)
Hence, the parallel use of accurate mass ES-MS and/or ES-MS2 was essential for unequivocal identification of AsIII–PCs as the example of the AsIII–PC3 complex in T. alata roots showed. The accurate mass using online the Orbitrap MS detected at the retention time of the As/S peak was an m/z of 844.0917, which is only 0.0011 Da (delta −1.06 mmu) different from the theoretical mass (m/z 844.0928) of AsIII–PC3. This accurate mass gave a good account of the elemental composition of the peptide but did not give the correct structure. For structural information ES-MS2 is necessary. The loss of glutamic acid in MS2 can give the first clue for the molecular structure. The use of HPLC–ICP–MS/ES-ion-trap MS gave sufficient structural information for identification of AsIII–PC3 (data not shown). However, it should be mentioned that this is sometimes not enough, as demonstrated in an earlier publication for the identification of the first pentavalent arsenic binding to peptides as dimethylarsinothioic glutathione complex in which 1H NMR of a standard was needed before unequivocal assignment could be made for the most abundant peptide of DMAV in a Brassica extract [27].
Species-independent quantification of AsIII–PC complexes
The use of ES-MS for quantification of AsIII–PC complexes was hampered by the fact that pure arsenite phytochelatin standards were not available and their formation by mixing phytochelatin(s) and arsenite is not quantitative, neither are the formed complexes stable. In addition, quantification using ES-MS is strongly matrix-dependent and is best done using the standard-addition approach. On-line coupling of reversed-phase HPLC with a methanol gradient run to ICP–MS led to the additional problem of changing sensitivity for arsenic and sulfur in dependence on the gradient. It has already been reported that the presence of methanol in the mobile phase has an effect on ICP–MS signal intensities [28, 29]. Hence the ICP–MS response for arsenic and sulfur had to be evaluated each time for different mobile phase compositions. In the ICP–qMS (xenon) setup enhancement of the arsenic and sulfur signals up to approximately 10% methanol was noted before their intensities slowly decreased (Fig. 6), which could also be confirmed by using the HR-ICP–MS. The reasons for the change in sensitivity are the same for all three instrumental setups. The initial enhancement of the signal intensities is caused by three mechanisms: (1) charge-transfer reaction from C+ species (derived from the increasing organic loading of the plasma) to analyte atoms; (2) improvement in the nebulisation transport (methanol = surfactant) of the sample; and (3) shift of the zone of maximum ion density in plasma [28, 29]. Local cooling of the plasma could have been the reason for the decrease of the signal intensities between 10 and 20% methanol, since the dissociation of the methanol requires energy which is taken from the plasma [28]. Using these species-independent methanol-adjusted standards As:S ratios, individual AsIII–PC complexes, and unidentified As peptides can be quantified without having the molecular-species as standards. The AsIII–PC complexes in twelve root extracts from twelve individual T. alata plants exposed to 1 mg AsV L−1 were quantified in order to measure biological variability and instrumental reproducibility of the AsIII–PC complexes. This is useful since in all experiments different plant extracts were used, because the AsIII–PC disintegrate over time even when stored at sub-zero temperature [30]. The root contained just below 10 μg As g−1 fresh weight, from which the majority could be extracted and successfully eluted from the column using formic acid–methanol as mobile phase. The major AsIII–PC complex was AsIII–PC3 followed by AsIII–PC4 and AsIII-(PC2)2 and additional unidentified As–peptides were also quantified (Table 2). An RSD between 36 and 52% for the individual AsIII–PC complexes indicated large biological variability or complex re-arrangements, whereas the overall variability of the sums of As peptide complexes seemed more reproducible (RSD 13%).

Effect of increasing methanol concentration in the mobile phase on the ICP–MS signal intensities of arsenic and sulfur using ICP–qMS with Xe as collision gas. As standards Methionine and DMAV were used as standards
Only with a very high degree of sophistication of the used analytical system was it possible to identify unambiguously arsenic peptides in the plant extract by using HPLC–ICP–MS and ES-MS. However, how can one prove whether, or not, the arsenic peptides have disintegrated or formed during the storage or extraction process, especially as extraction is known to be the hardest step to control in respect of species integrity during speciation analysis. Here cytosol of plant cells is mixed with vacuoles and cell membranes in an abiotic extractant, a scenario which can significantly change the chemistry of unstable arsenic peptides. The only way to control this step is to use a complementary method which does not need this extraction step: X-ray absorption spectrometry.
XANES and EXAFS
XANES and EXAFS can be used to predict the oxidation state and the bond length to the nearest ligands of the element in question. Although, these techniques do not provide full molecular information, and suffer from high detection limits, their advantage is that they can be applied directly to the fresh plant root. Eight scans of 45 min each had to be accumulated and summed in order to collect data with signal-to-noise levels good enough for EXAFS analysis, due to the low concentration of arsenic in the samples (<10 mg kg−1 fresh weight), although fewer scans would have sufficed for just the XANES data (Figs. 7 and 8). It was apparent from the XANES data that the arsenic in the roots of the exposed T. alata did occur in different chemical environments, indicated by the two different maxima (Fig. 7). For XANES and EXAFS the data were collected at the As K edge. The XANES data have been fit with the four model compounds to give the lowest fit-index (0.72%). Fit index of the calculated XANES spectra with experimental XANES spectra is defined as \( {\sum {{{\left[ {{\left( {I_{{obs}} - I_{{calc}} } \right)}^{2} } \right]}} \mathord{\left/ {\vphantom {{{\left[ {{\left( {I_{{obs}} - I_{{calc}} } \right)}^{2} } \right]}} n}} \right. \kern-\nulldelimiterspace} n} } \) where n is the number of points in each spectrum. The XANES data showed that a significant amount of arsenic was chemically similar to the AsIII–(GS)3 standard (53%), i.e. bound to three S and trivalent, while some arsenic was bound to oxygen and both trivalent (38%) and pentavalent (9%). DMAV contribution did not improve the fit index, and could be considered not to be present in significant amounts in the plant root. The absence of DMAV has also been confirmed by anion-exchange chromatography (data not shown), which excluded in-planta methylation of AsV. The distribution of the other species indicated that most of the absorbed AsV was transformed; either to arsenite or to trivalent arsenic bound to the thiols of peptides. This was in agreement with uptake and transformation processes seen in other organisms [31]. The EXAFS data were very noisy (R-factor of 44) and were subject to a large error for the fit, as indicated in Fig. 8. The EXAFS data, however (Table 3), showed contributions from AsIII–O at 1.75 Å and AsIII–S at 2.27 Å, consistent with the XANES fitting.

XANES measurements of the roots of freshly exposed T. alata compared with aqueous standards used for the fit. The adsorption edge at 11.873 eV is characteristic of trivalent arsenic bound to three sulfur ligands, while the adsorption edge at 11.877 eV characterizes pentavalent arsenic bound to four oxygen atoms

EXAFS from the roots of a Thunbergia alata, exposed to 1 mg L−1 As, as arsenate, 24 h, after rinsing with phosphoric acid. The fits indicate that arsenic binds to two different ligands. The bond length of 2.27 ? indicates that trivalent arsenic binds to reduced sulfur as in glutathione whereas 1.75 ? is characteristic of an arsenic–oxygen bond
This 53% of arsenic binding to S of peptides of the one plant was at the lower end but within the range of the twelve different root extracts measured using HPLC–ICP–MS (55–64%) (Table 2). It should be considered here that the plant was stressed because of transport from Aberdeen to the CCLRC lab, which may have affected the formation of peptides in the plant. Considering the methodology errors of both methods and the biological variability, the XANES/EXAFS results confirmed the arsenic speciation revealed by use of HPLC–ICP–MS/ES-MS. This showed that the arsenic–sulfur bond was not formed during extraction or chromatography, but existed in the original plant, most likely in the vacuoles. Hence, the extraction procedure, although not very gentle or even under physiologically identical conditions, preserved the arsenic peptide bond and did not create new ones.
Conclusion
It was confirmed by XANES/EXAFS that formic acid extraction can be used for detection and quantification of arsenic peptides in plants using HPLC–ICP–MS. Hence, we can trust the mass spectrometric analysis under certain conditions. Xe as a collision gas for ICP–qMS was not quite good enough for monitoring sulfur-containing peptides in real samples. Although the sensitivity of HR-ICP–MS was an order of magnitude better, the As:S ratio as an indicator of arseno-peptide complexes could only be used for the major AsIII–phytochelatin complexes. The differences between the experimentally obtained and theoretically expected As:S ratios made clear that one can not rely on the ICP–MS data concerning the identification of AsIII–PCs using the As:S ratio and their quantification on the basis of sulfur, which is, in both cases, attributed to the high detection limit for sulfur. However, it should be pointed out that the As:S ratio was sometimes misleading since the chromatographic resolution of the current chromatographic method was not sufficient for the diversity of arseno-peptides in plants. It was shown that, in any case, molecular mass spectrometry (ES-MS accurate mass or ion-trap) in dual mode with ICP–MS was necessary to identify and quantify the arsenic species in the extract. The quantification of free reduced and oxidised phytochelatins in plant extracts using ICP–MS remains still a challenge.
Notes
Here, the term “organo-arsenic species” is not only restricted to compounds with As–C bonds but also to complexes with organic ligands and arsenic as central atom, for example AsIII–PCs.
References
Francesconi KA, Kuehnelt D (2004) Analyst 129:373–395
Hansen HR, Jaspars M, Feldmann J (2004) Analyst 129:1058–1064
Kahn M, Raml R, Schmeisser E, Vallant B, Francesconi KA, Goessler W (2005) Environ Chem 2:171–176
Nischwitz V, Kanaki K, Pergantis SA (2006) J Anal At Spectrom 21:33–40
Hansen HR, Pickford R, Thomas-Oates J, Jaspars M, Feldmann J (2004) Angew Ch Int Ed 43:337–340
Raab A, Schat H, Meharg AA, Feldmann J (2005) New Phytol 168:551–558
Raab A, Meharg AA, Jaspars M, Genney DR, Feldmann J (2004) J Anal At Spectrom 19:183–190
Rauser WE (1995) Plant Physiol 109:1141–1149
Zenk MH (1996) Gene 179:21–30
Cobett SC (2000) Plant Physiol 123:825–832
Meharg AA, Hartley-Whitaker J (2002) New Phytol 154:29–43
Quaghebeur M, Rengel Z (2005) Microchim Acta 151:141–152
Raab A, Ferreira K, Meharg AA, Feldmann J (2007) J Exp Bot 58:1333–1338
Rosenberg E (2003) J Chromatogr A 1000:841–889
Meharg AA, MacNair MR (1992) J Exp Bot 43:519–524
Sheldrick WS, Häusler HJ (1987) Z Anorg Allg Chem 549:177
Schneidersmann C, Hoppe R (1991) Z Anorg Allg Chem 605:67
Trotter J, Zobel T (1995) J Chem Soc 4466
Peters K, Peters EM, von Schnering HG, Wojnowski W, Tamulewicz S, Adacki KR (1997) Z Kristallogr New Cryst Struct 212:343
Bandura DR, Baranov VI, Tanner SD (2001) Fresenius J Anal Chem 370:454–470
Tanner SD, Baranov VI, Bandura DR (2002) Spectrochim Acta B 57:1361–1452
Douglas DJ (1997) J Am Soc Mass Spectrom 9:101–113
Marcus RK (2004) J Anal At Spectrom 19:591–599
Rowan JT, Houk RS (1989) Appl Spectrosc 43:976–980
Wallschläger D, Stadey CJ (2007) Anal Chem 79:3873–3880
Raab A, Feldmann J, Meharg AA (2004) Plant Physiol 134:1113–1122
Raab A, Wright S, Jaspars M, Meharg AA, Feldmann J (2007) Angew Ch Int Ed 46:2594–2597
Hu Z, Hu S, Gao S, Liu Y, Lin S (2004) Spectrochim Acta B 59:1463–1470
Larsen EH, Stürup S (1994) J Anal At Spectrom 9:1099–1105
Bluemlein K, Raab A, Feldmann J (2008) in preparation
Mukhopadhyay R, Rosen BP, Silver S (2002) FEMS Microbiol Rev 26:311–325
Acknowledgements
This work has been supported by NERC (NE/B505789/1). Many thanks to F. Bahrami (STFC Daresbury Laboratory, UK) and Julian Wills (ThermoFisher, Bremen, Germany) for their assistance in using their instrumentation. KB thanks the College of Physical Sciences for her scholarship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bluemlein, K., Raab, A., Meharg, A.A. et al. Can we trust mass spectrometry for determination of arsenic peptides in plants: comparison of LC–ICP–MS and LC–ES-MS/ICP–MS with XANES/EXAFS in analysis of Thunbergia alata . Anal Bioanal Chem 390, 1739–1751 (2008). https://doi.org/10.1007/s00216-007-1724-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-007-1724-y