
Abstract
Recent developments of element-specific microscopy techniques using synchrotron radiation are opening new opportunities for the analytical investigation of various heterogeneous materials. This article provides a general description of the operational principles of different microscopes allowing chemical and structural imaging combined with micro-spot spectroscopic analysis. Several selected examples are used to illustrate the potential of the synchrotron-based methods in terms of imaging and chemical sensitivity for identification of spatial variations in the composition of morphologically complex and nano-structured inorganic and organic materials, including biological samples.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most materials are heterogeneous and complex by nature or design and one of the challenges in their characterization is to combine at the same length scales morphological studies with methods providing quantitative information about the chemical composition. The recent progress in development of instrumentation for probing the properties of matter at sub-micrometer scales fulfills the demand of many important fields, from nano-technology, e.g. for characterization of nano-materials and devices, to bio-medical research, e.g. for understanding the functions of cells, bone tissue and sub-cellular structures.
The chemical information for the specimen can be obtained by using a variety of spectroscopic methods based on the excitation processes in the matter as a result of interaction with electromagnetic radiation. The spectral features are obtained by monitoring photon absorption and photon or electron emission of specimen irradiated by properly tailored photon beams with selected wavelengths. Due to the higher penetration depth of X-rays as compared to electrons, the photon-in/photon-out spectroscopy methods are mostly bulk sensitive and can be used for characterization of both conductive and non-conductive materials, whereas the photon-in/electron-out spectroscopy methods are surface sensitive and not applicable to insulators. The exploitation of these methods for microscopic analysis and imaging with high spatial resolution requires sufficiently intense photon beams. The great advantage of the synchrotron photon sources is that they provide not only very bright but also polarized and tunable radiation with wavelengths from micrometers (far-infrared) to Angstroms (hard X-rays) [1]. This has opened the opportunity to build various types of microscopes implementing the well-known spectroscopic techniques for providing chemically specific information.
The complementary capabilities of different microscopy approaches in terms of imaging, spectroscopy, spatial and time resolution are strongly requested by the multi-disciplinary research programs at the synchrotron facilities and have motivated continuous investments in development of instrumentation for imaging with spectroscopic analysis. This article provides an overview of the possible chemical imaging approaches offered by the various microscopes at synchrotron radiation facilities. Using selected examples we will illustrate their potential for bio-medical and material research, in terms of combined chemical and morphological information. Most of the results were obtained in the authors’ institution, which has several beamlines hosting different microscopes.
Synchrotron-based microscopes: working principles, advantages and limitations
The chemical imaging considered in this paper is based on the following spectroscopic methods: X-ray absorption spectroscopy (XAS), infrared (vibrational) spectroscopy (IR), X-ray fluorescence spectroscopy (XFS) and X-ray photoelectron spectroscopy (XPS). XAS and in particular X-ray absorption near-edge spectroscopy (XANES) monitors the transmitted photons or the total emitted electron or photon signal while varying the energy of the incident photon beam [2]. The elemental sensitivity of absorption spectra comes from the characteristic binding energies of core electrons, ranging from a few tens to a few hundred thousand electronvolts, which give rise to X-ray absorption thresholds and resonant electronic transitions from the core levels into unfilled electronic states, governed by well-established selection rules [2]. Using polarized photon beams one can probe the absorption of light with circular (left and right) or linear polarization at the absorption edges of the constituent atoms (X-ray magnetic circular or linear dichroism (XMCD or XMLD), which adds information on the average magnetic moment and symmetry of the chemical bonds. IR is a widely used technique monitoring the vibrations of chemical functional groups as a result of absorption of infrared radiation in a specific wavelength range, regardless of the structure of the rest of the molecule. The major benefit of using the IR range of synchrotron radiation is in spectromicroscopy, because the higher brightness compared to globar sources allows lateral resolution at the diffraction limit (determined by the IR wavelength) to be achieved [3]. XFS detects and energy filters the photons emitted as a result of secondary de-excitation processes occurring in the atoms interacting with monochromatized X-rays [4]. Since the X-ray probe offers orders of magnitude higher sensitivity than electron probes, the element-specific XFS using synchrotron light has become powerful tool for quantitative trace element analysis, important for understanding material properties such as second-phase particles, interfacial segregation and contaminations. XPS detects and energy filters the electrons emitted from atomic core and valence levels as a result of interaction with monochromatic photons [5]. From all emitted electrons, those from the atomic core levels provide the most straightforward quantitative compositional information and also are excellent fingerprints for the chemical state and/or structural and chemical environment of the emitting atoms. XPS is a surface-sensitive technique and is very appropriate for investigations of interfacial phenomena: the sample probing depth is determined by the kinetic energy of the photo-emitted electrons and can vary from less than ten to a few hundred Ångströms.
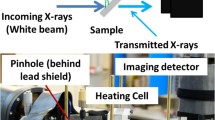
In brief, the potential and the applications of the microscopes are determined by their operational principle and the contrast mechanisms. The spatial resolution and image formation are achieved by two different approaches, which classify the instruments as full-field imaging and scanning (see Fig. 1) [6–10].

Schematic of the full-field imaging and scanning-type microscopes
In the full-field imaging microscopes the incident photon beam is first demagnified to micrometer dimensions to illuminate homogeneously a selected area of the sample, using diffractive condenser, capillaries or a focussing mirror. The magnified image of the irradiated area is obtained by projection of transmitted photons or emitted photoelectrons. The instruments, using projection of transmitted photons, are called transmission X-ray microscopes (TXM). They work similarly to a visible light microscope: an objective lens illuminates the specimen, and a second objective lens (Fresnel zone plate (ZP) lenses) behind the specimen generates a magnified image onto a spatially resolving detector [7, 8]. The instruments using projection of emitted electrons are called X-ray photoemission electron microscopes (XPEEM) [8]. The emitted electrons are collected by an objective lens, and by means of electrostatic and/or magnetic lenses a magnified image of the probed area is projected onto a screen, converting the electron signal into visible light. These instruments can use both XANES and XPS for chemical information and are a special category not further considered in the present paper.
The scanning approach uses a microprobe and an image is acquired in transmission or emission modes by detecting the photon or electron signal by raster-scanning the sample. The microprobe in the scanning X-ray microscopes is formed using reflective (glancing or near-normal-incidence mirror systems) or diffractive (ZP lenses) optical elements [1, 11].
In the transmission-type microscopes, as in visible light or transmission electron microscopes, versatile imaging contrast mechanisms can be used for morphological studies. They are based either on attenuation or phase shift of the X-ray beam by the sample. Absorption contrast is usually used to map the sample density. Phase-contrast imaging (differential phase contrast, differential interference contract, dark field imaging etc.) is more sensitive to the internal structure, even when absorption is low, and can be further enhanced by tuning the X-ray energy [12]. Particular elemental constituents can be located using measurements on each side of the absorption edges, giving an element-specific difference image. By scanning the X-ray beam’s energy across the absorption edge of an element information on its chemical state (XANES) can be provided.
The full-field imaging approach, TXM, more easily achieves high spatial resolution, because of the static mount of the sample during data acquisition and the ease of applying all possible phase contrast modes [13]. Another advantage of the ‘one shot’ TXM is the short acquisition time (down to sub-second range at present for an image), which makes the approach very suitable for dynamic studies and for obtaining 3D structures (X-ray tomography). For 3D imaging the sample should be mounted into a rotating stage and the 2D projection of the structures is viewed at different rotation angles. The series of X-ray images, recorded at different angles, can be reconstructed in the computer to give the full 3D information [14]. The disadvantage compared to scanning microscopes is that the TXM is adequate only for XANES: the micro-spot spectroscopic analysis is slow, because it requires the recording of stacks of subsequent images at energies across the selected absorption edge. The scanning approach offers maximum flexibility to use different photon and/or electron detectors for spectroscopic micro-spot analysis and imaging. The use of multi-element detectors allows different forms of image contrast to be produced by suitable combinations of the various signals. In principle scanning-type microscopes can work both as a scanning transmission X-ray microscope (STXM) with XANES or a scanning photoemission microscope (SPEM) with XANES complemented with XFS (using hard X-rays) [15] or PES (using soft X-rays) [10].
Here we would like to stress that in terms of imaging resolution, X-ray microscopy approaches lie between electron and light microscopy and have comparable or better resolution than other charge-particle methods, such as particle-induced X-ray emission (PIXE) and secondary ion mass spectrometry (SIMS) [16]. In fact, using photon optics we cannot break the frontier of nearly atomic resolution achievable by transmission electron microscopes (TEM), but using tunable synchrotron X-ray radiation has several advantages compared to microscopy with charged-particle probes, as summarized below.
X-ray microscopy is quantitative and less invasive, having enhanced ability for in situ studies of dynamic processes. TXM and STXM have relative design simplicity and the ability to penetrate inside thicker samples in a near-native environment, delivering high-contrast images with elemental information and time resolution. The most distinct advantage is in the variety of spectroscopic approaches, described above. Compared to TEM-EELS, the advantages of XANES in detecting functional groups in organic and polymer samples have been well recognised [17]. In SEM the interaction process of the impinging electrons, which leads to a large interaction volume, prevents the XRF compositional analysis at very high spatial resolution. Another important distinction from charged-particle probes is that X-rays do not interact with applied electric or magnetic fields and both linearly and circularly polarized X-rays are available. Contrast due to polarization is invaluable in distinguishing fluorescence and diffraction signals, and determining orientation of chemical moieties or magnetic domain structures at high spatial resolution, using XMCD or XMLD.
In the IR microscopes the incident beam is both demagnified and delimited by an aperture in an optical plane conjugated to that of the sample, the aperture size determining the portion of the sample probed by the IR beam [3]. Confocal IR spectromicroscopy, realised by the presence of pinholes which allow only light from the plane of focus to reach the detector, can be performed in both transmission and reflective mode. Mapping of selected IR spectral features is performed by raster-scanning the specimen under the microscope objective and using absorption values at specific frequencies to generate image contrast. Because of the limited brightness of the globar source there is a dramatic loss of intensity when closing the apertures below 20 μm, resulting in a poor signal-to-noise ratio and long collection times. The high brightness of a synchrotron source allows retention of a relatively high throughput even with apertures closed to a size comparable to the IR diffraction limit (e.g. 1.7 μm at 4,000 cm−1). At such small apertures the signal-to-noise level obtained with a synchrotron source is typically three orders of magnitude higher and the time for mapping is correspondingly shorter.
Imaging of biological specimen using X-ray transmission microscopy
Transmission X-ray microscopy has found a lot of applications in analyzing the morphological structure of specimen at the sub-100-nm length scale and extracting the chemical information by means of XANES. The higher penetration depth of X-rays compared to charged particles allows imaging of complete cellular and sub-cellular structures in their natural environment without slicing [18]. Similar to scanning or transmission electron microscopes, the high radiation dose necessary to image very small features with a decent signal-to-noise ratio requires in many cases fixing or cryogenic freezing of organic matter and use of the most appropriate contrast techniques for imaging.
Recently using a novel approach the STXM and TXM have been integrated into a single instrument with fast switching between the two microscopy modes, called TwinMic [19]. By preserving the performance of each microscopy approach, it has opened an opportunity to perform fast imaging for dynamic studies with TXM, combined with STXM and micro-spectroscopy (XANES). In the near future, the implementation of XFS will also allow the STXM module to work as a SPEM.
The imaging potential of the TXM module in TwinMic for resolving features in cells is illustrated by the images of U2OS 2e11 human cells [20], shown in Fig. 2. The eventual aim of the project is to develop novel genetically encoded contrast labels for the detection and reporting of sub-cellular processes, such as HIV-1 transcription, through specific targeting of externally administered probes.

X-ray micrographs of the cell nucleus and cytoplasm of HOS 2e11 cells, as indicated by the cell scheme in the middle. In the left image the lower part is the cell nucleus, where the three highest absorbing zones (the darkest) are the cell nucleoli, whereas the upper part is the cytoplasm. The right image shows the cytoplasm with part of the cell nucleus on the left side. The images were acquired with photon energy of 720 eV using absorption contrast. The exposure time was 10 s. The HOS 2e11 cells were grown to confluence on silicon nitride membranes and subsequently fixed with fresh 3.7% buffered paraformaldehyde. Samples were then dehydrated by increasing ethanol concentration
Figure 3 illustrates the chemical imaging ability of TXM mode. The high resolution images of a soil sample are taken below and above the L-absorption edge of iron. The Fe ions are known to act as coagulation agents promoting the flocks or aggregates of humic substances in the soil, which play an important role in environmental processes. These studies are aimed at visualizing the distribution and size of the iron-rich particles. The final goal is to determine the morphology of organic matter as a function of concentration and chemical state of iron, which has direct impact on the bioactivity and fertility of the soil [21].

Morphology and distribution of hematite aggregates in a soil sample, dried from its aqueous dispersion medium. The images were measured at different photon energies across the Fe L-absorption edges. The X-ray absorption image taken below the Fe L-edge (703 eV) demonstrates the morphology of the sample. The Fe-rich areas (best seen in the indicated yellow zones) are revealed by imaging with photon energy around the Fe-L edge (707 and 703 eV)
One of the major advantages of the scanning mode is that different signals from one or multiple detectors can be acquired simultaneously. Figure 4 illustrates a simultaneously detected absorption image (left) and the differential phase contrast (DPC) image (right) obtained by using a configured detector in TwinMic scanning mode [22]. The samples are marine planktonic diatoms Coscinodiscus sp., which are encased in an intricately patterned biosilicate skeleton. The higher penetration power of X-rays helped to reveal that the ornate biosilicate shell consists in some cases of a double hexagonal net, not observed with TEM. The DPC principle applied in the scanning microscope is detection of local phase gradients through the small phase shift between two close points of the sample [23, 24]. Phase information is extracted by calculating the first moment derivation of the scattered light. The DPC mode can lead to an increase in image contrast of low absorbing specimens, such as cells, by up to 20%.

X-ray micrographs of marine planktonic diatoms Coscinodiscus sp. acquired simultaneously with a configured detector in scanning mode: left absorption contrast, right differential phase contrast. The images were acquired with 720-eV photon energy and dwell time of 80 ms per pixel
Imaging and micro-spectroscopy of bone specimens using scanning microscopy in X-ray transmission and emission modes
Implementation of a fluorescence X-ray detector in scanning microscopes and using proper X-ray wavelengths allow micro-XFS and micro-XANES with resolution determined by the size of the microprobe, provided the probed sample has a uniform thickness and negligible density variations. The potential of these instruments will be illustrated by some recent results, focussed on determination of the components and their distribution within a bone tissue.
Bone mineral consists of apatite-like nanocrystallites, a non-stoichiometric calcium phosphate phase acting as an ion reservoir. Numerous minor and trace elements (Na, Mg, Cl, Sr, Mn, etc.) are present as well. Investigation of the bone mineral content at a microscopic scale is of particular interest, since it can give new insights into remodelling activities, mineralization processes, effect of drugs and related mechanical properties. Diseases and pathological conditions cause changes in bone tissue affecting its properties, morphology and bone turnover rate. The images in Fig. 5 of a 10-μm-thick transversal slice of chicken bone embryo (20 days old) represent one of a series at different stages of maturation. They are obtained in transmission (absorption contrast) and fluorescence spectroscopy modes and illustrate the microscopic bone morphology and the lateral distribution of Ca and P. Using XRF enabled simultaneous mapping of the distribution of different elements eventually present inside the bone with sensitivity (a few parts per million). Figure 5a reveals the bone architecture, composed by sub-structures with some specific orientation. These substructures are composed of small nanometre-sized crystallites stacking on the collagen fibres. As expected, the fluorescence maps obtained by tuning the fluorescence detector to the Ca or P emission lines are almost identical showing perfect co-localization of Ca and P. However, the bone morphology and stoichiometry (Ca/P ratio) provide relevant biological information for the maturation stage.

Images of a chicken bone embryo specimen: a large-scale transmission image, taken with step size 10 m and integration time 100 ms illustrating the bone morphology (b) and c phosphorus and calcium fluorescent images of the square area indicated in a taken with step size 0.5 μm and integration time 1 s. Photon energy = 4.1 keV. Sampling points are numbered
The micro-XFS spectra acquired in selected 0.25-μm2 sample spots provide detailed quantitative information about the local elemental composition, which was elucidated by measuring reference spectra of samples with known elemental concentration. The spectrum in Fig. 6a, taken from point 1, shows the presence of Ca, P, K and Cl, the last two elements being related to the bone soft tissue. The presence of only the main bone mineralization components indicates that at this very initial ‘embryo’ stage the other elements are either below the detection limit or not yet incorporated inside the bone mineral [25].

a X-ray fluorescence spectra of a bone sample taken at point 1, indicated in Fig. 5b, taken with photon energy of 4.1 keV. b Spatially resolved at the Ca K-edge taken at the points 1 and 2, indicated in Fig. 5b. c XANES spectra at the Ca K-edge of apatite calcium phosphates: S-HA is stoichiometric hydroxyapatite; PCA are poorly crystalline apatites at different maturation stages (0, 3 days, 16 months). d Derivatives of the XANES spectra from the series of spectra in c. The micro-XANES is measured in transmission. The most important features are indicated a–c; see Text for details
Bone mineral crystals are in metabolic interrelation with the body fluids, changing in size, chemical composition and stoichiometry with age, disease and chemical treatment. There are also biological aspects, due to continuous bone turnover, i.e. formation of a new bone, which contains the youngest crystals and resorption of bone, which contains crystals of various ages including the oldest ones. Thus crystals from all stages of maturation should be present in the bone sample and being non-stoichiometric apatite phases they differ by the local environment of the Ca ion. As several biological properties of bone are related to these environments, like homeostasis, ion exchange or protein attachment, it is important to access the highly reactive calcium sites of bone by a direct spectroscopic method. The complementary micro-XANES analysis, based on monitoring the total emitted fluorescence signal (or transmitted X-rays in the case of thin enough samples) while scanning the X-ray energy across the selected core electron absorption edge, adds information about the chemical environment of the Ca ion. This information is of fundamental importance for understanding the process of bone mineralization during maturation process and is obtained by comparing the micro-XANES taken at points 1 and 2 (Fig. 6b) with those measured for synthesized non-stoichiometric apatite analogues of the bone mineral (PCA). The latter have the same physico-chemical properties as the bone mineral during maturation and are excellent references for the different types of non-apatite environments of the calcium ion [26, 27]. The spectra of the PCA analogues at different maturation stages, shown in Fig. 6c, exhibit very similar features, the most important indicated as ‘a’, ‘b’ and ‘c’. Note that the pre-edge energy position and intensity are independent of the maturation state of our PCA, suggesting that both the effective charge and the site of symmetry of the Ca atom remain unaltered. As the maturation time increases, the dashed line ‘a’ becomes a bit narrower: this is assigned to the existence of two calcium sites with different coordination [28]; the structures ‘b’ at 4,056 and 4,063 eV become more pronounced and the ‘c’ maximum shifts continuously from 4,096.2 to 4,098.1 eV, as can be seen clearly in the derivatives of the spectra, shown in the Fig. 6d. The energy shifts of structure ‘c’ reflect the changes in the Ca–O bond length, becoming shorter with increasing the maturation time. The μ-XANES from the specific points (1, 2) are very similar to those obtained for the calcium phosphate analogous to bone mineral, revealing prevalence of non-apatite micro-phases in the sample. We would like to note that for the first time biologically reactive non-apatitic environments of the calcium ion have been evidenced by a direct spectroscopic method. This is essential to understand the path of bone maturation and reactivity and relates to elucidation of the effect of drugs or the alterations induced by pathology.
Chemical imaging and micro-spectroscopy using scanning microscopy in XPS mode
The intensity and kinetic energy of the electrons emitted from a specific core or valence level of the elements provide information about the local elemental concentration and chemical state of the constituents at the sample surface and near-subsurface region. The core level energy shifts and changes in the valence spectra due to interactions with other atoms or molecules are fingerprints of the actual chemical state and electronic structure, band-bending and local charging. The use of XPS as a spectroscopic method limits the SPEM applications to studies of specimen in solid state and in ultra-high vacuum environment (pressures lower than 10−6 Pa), but on the other hand chemical imaging with μ-XPS is the best analytical method for probing surface and interfacial phenomena at sub-micrometer scales.
Among the recent achievements in XPS-based characterization of nano-structured materials are the results obtained with C- and MoSx-based nanotubes. Due to their low dimensionality and high anisotropy the nanotubes exhibit a large variety of mechanical and electronic properties, which makes them an excellent material for nano-technological applications. The high spatial resolution of SPEM allowed us to localize the nanotube bundles (approximately 5- to 10-μm long and up to 0.5-μm wide) and select the areas for systematic spectroscopic measurements along the nanotubes axes. An important finding in these studies is the position dependence of the photoemission valence and core level spectra of aligned nanotubes, reflecting the different structural organization along the wall and the tip of the nanotubes [29, 30]. Figure 7 shows a typical cross sectional Mo 3d image of aligned MoSx-based nanotube bunches. The difference between the S 2p spectra taken at the tips, the sidewalls and the base (interface with substrate on which are grown) reflect the position and size dependence of the local chemical and electronic structure. There is significant difference compared to the S 2p spectra of the reference MoS2 crystal as well.

Left Mo 3d image (10 × 10 μm2) of aligned MoSx nanotubes grown on Si substrate. Right S 2p spectra taken at different parts along the nanotube bundles. The fitting components indicate different bonding configurations of S within the nanotube structure. The bottom S 2p spectrum is of the MoS2 compound
Monitoring degradation processes in electronic devices and understanding the impact of the surface morphology on the properties metal/semiconductor interfaces is imposed by the stringent technological requirements for realization of reproducible and thermally stable ohmic contacts. The great advantage of using the XPS method in SPEM studies is that it allows the probing of both the local composition of the interfacial region and the corresponding band bending (BB) at the surface.
Extensive SPEM studies of the metal/GaN interfacial interactions revealed that the observed lateral variations in the composition were most often induced by morphological and crystallographic defects of the GaN epilayers [31–33]. The ‘defect’ areas, identified by SEM prior to inserting the GaN sample into the SPEM, were also located in the Ga 3d maps taken before Ni deposition. Figure 8 shows Ni 3p and Ga 3d images illustrating the chemical heterogeneity developed in a defect region at the Ni/GaN interface after thermal treatment. Ni alloys with Ga and forms gallides and the best fingerprint for the local reactivity is the amount of the released Ga, which appears as a new component, R-Ga, in the Ga 3d spectra. Depending on the temperature the gallides at the interface change their stoichiometry, best reflected by the binding energy and relative weight of the R-Ga component in the Ga 3d spectra. The images in Fig. 8 clearly show that the most rigorous interfacial reaction has occurred at the defect sites, which also become enriched with Ni.

Top left images of the total Ni 3p and Ga 3d core level emission taken in a defect area of Ni/GaN contact; bottom left Ga 3d images of the emission in the energy windows corresponding to the R-Ga and GaN components, as indicated in the panel with Ga 3d spectra; right Ga 3d spectra of GaN substrate (bottom) and of the defect and non-defect regions of the Ni/GaN contact where compositional inhomogeneity has developed upon heating to 200 °C. The R-Ga component corresponding to the NiGa alloy is plotted as well. BB indicates the band bending shift after metal deposition
Another successful study with SPEM helped in understanding the degradation mechanisms leading to appearance of dark spots on the cathode surface of organic light emission devices (OLEDs) [34]. The elemental maps and micro-spot XPS spectra in Fig. 8 illustrate the changes in morphology and chemical composition of damaged Al cathode surface OLEDs that have undergone degradation during operation. The created fractures appear as dark holes in the Al 2p image because there is no Al 2p emission from the disrupted areas. Depending on the advancement of the degradation process the organic layers or the InSn oxide (ITO) anode below are exposed. The central part of the fracture appears very bright in the In 3d map, whereas it has low intensity in the C 1s map, which indicates that the organic layer has also been locally damaged, exposing the ITO surface below. Another distinct feature of the In 3d maps is the presence of In-containing species on the Al surface around the fractures, the amount of these species decreasing gradually moving away from the hole (see the In 3d concentration profile below the In image in Fig. 9). This is direct evidence that the degradation also involves the ITO anode and a fraction of generated In-containing volatile species is deposited on the Al anode around the hole. Similar contrast is observed in the C 1s maps, which indicates co-deposition of organic species, released from the locally decomposed organic layers. More detailed information about the chemical composition within the fractured area and the surrounding cathode surface were provided by the μ-XPS spectra in Fig. 9 measured in the different regions. The spectra, measured in the dark holes are very similar to the ITO spectra, except the additional new Si 2p line, originating from the ITO glass support and indicating that within the damaged area the thickness of the ITO has been dramatically reduced due to material loss. The very small C 1s peak in the spectra confirms that locally the organic layer has also been removed. Comparing the μ-XPS spectra from the cathode areas in the vicinity and away from the fracture confirm the deposition of the ITO constituents, In and small amount of Sn, and C-containing species from the organic layers. The similar results obtained for different ambient and operating conditions led to the conclusion that the lateral inhomogeneities in the surface topography and chemistry of the ITO films should be one of the major reasons for electric spikes causing the cathode disruption and device failures.

Top Al 2p, C 1s and In 3d maps in a damaged region of the Al cathode, (a ‘dark’ spot in the OLEDs). Since the deposited In film screens the Al 2p emission these parts appear darker in the Al 2p map. The concentration profile of In deposited on the Al cathode surface around the hole in shown in the right panel. Bottom XPS spectra taken inside the hole (1), on the Al cathode in the vicinity of (2) and far away (3) from the hole. The top spectrum, corresponding to the anode (ITO), is shown for the sake of comparison with spectrum 1
Chemical imaging and micro-spectroscopy using synchrotron-based IR spectro-microscopy
Although IR microscopy has modest spatial resolution compared to X-ray microscopy the greatest advantage is the combination of high chemical sensitivity and the non-destructive character of infrared radiation. Being a photon-based technique it can be used for characterization of non-conductive materials and is very adequate for studies of organic materials and living objects.
The first example illustrates the application of the IR microscopy as a complementary technique in characterization of the quality of encapsulation of the OLED devices, namely the protective layer against degradation due to contact with moisture or other active gases in the air. This protection is a critical step in the OLEDs fabrication and one of the solutions is deposition of organic and inorganic (oxide) films with thickness of the order of a few nanometres on top of the Al anode. The stability and duration of this protective ‘cap’ depend exclusively on the lateral homogeneity of the films, which requires very accurate optimisation of the deposition process parameters. The appearance of defects in the encapsulating layer, seen in the light microscope as bubbles of sizes up to a few hundred micrometers, is supposed to be a result of the non-uniformity in the deposited inorganic and organic films. Each of these barrier films has distinct IR spectral features, which allowed us to probe their lateral uniformity and correlate it to the defects observed in the light microscopes. We were able to detect the ‘bubbles’ of the protective layer in the IR reflectivity maps (Fig. 10a), because the local refractive index depends on the local chemical state of the Al anode surface. As can be expected, the Al cathode undergoes fast oxidation in defected regions because it has been exposed to air and humidity, and the locally oxidized surface has much lower reflectivity (the two bright areas in Fig. 10a). The images in Fig. 10(b,c) are IR absorption maps at 1,250 cm−1, corresponding to the –O–CH3 groups (b) of the organic layer and at 1,050 cm−1, corresponding to the silicon oxide contained in the inorganic layer (c). The contrast variations in the maps in Fig. 9(b,c) are very different, in particular in the defect areas. The variations in the relative abundance of organic component (b) appear almost unaffected by the presence of the defect, with the only exception in the middle of the larger defect area, where apparently the bubble has burst. The inorganic component (c) displays stronger non-homogeneous distribution with a reduced intensity in the regions where the bubbles are located. This supposes that a possible cause for defect creation is a local deficiency of inorganic film, which requires thorough examination of the Al cathode surface quality and deposition cycles.

Left map of the total light intensity reflected by the Al cathode, middle map of the 1,250 cm−1 absorption band corresponding to the organic layer, right map of the 1,050 cm−1 absorption band corresponding to the inorganic layer
IR spectromicroscopy on single living cells, or even at the sub-cellular level, has been made possible specifically by the application of synchrotron light [35]. Eukaryotic cells commonly have dimensions in the 10-μm range, too small for most spectromicroscopy and mapping experiments when using a globar source. Figure 11 illustrates the possibility to image the composition of sub-cellular structures in an intact cell. Mapping was performed by scanning the sample with a 3-μm confocal aperture, which is not possible using a globar source. The contrast in the IR images is generated by mapping the absorption bands characteristic of amide (a) and methyl/methylene (b) groups throughout the sample, fingerprints of the local content of protein and lipids, respectively. In the light microscope image of the same cell the nucleus is clearly visible in the lower right of the cell, together with other organelles. Comparison of the IR and light microscope images clearly demonstrates the differences in the distribution of proteins and lipids within the cell which can be correlated with cell structure.

IR images of a amide band absorption, b methyl/methylene band absorption and c the corresponding visible light image
Conclusions
The present microscopy instruments using synchrotron light and different contrast mechanisms have achieved their state-of-the art performance in exploration of heterogeneous phenomena at mesoscopic length scales, with increasing impact in micro-characterization of solid and liquid matter. The near-future upgrades aim at improving the lateral resolution, spectral resolution and data acquisition velocity. The progress is directly linked to further development of efficient, high resolution X-ray focussing optics for scanning and full-field imaging X-ray microscopes in order to achieve in all instruments resolution better than 25–30 nm, improving the transmission of the electron energy filters and the efficiency of the X-ray and electron detectors in order to work with better spectral resolution. The introduction of two-dimensional focal plane array (FPA) detectors in IR microscopy has allowed rapid imaging of the sample by forming the full infrared light image on the detector array and collecting the spectrum of each pixel. Imaging with FPA detector using synchrotron IR source is limited by the small size of the synchrotron spot projected onto the FPA plane. However, as reported recently, an FPA detector adequately filled with synchrotron IR delivers an excellent signal-to-noise ratio [36, 37]. An optical configuration will be shortly implemented on a permanent basis at SRC with the construction of a new dedicated beamline, where the FPA detector can be completely filled by proper tailoring the IR emission. Further steps forward are construction of ultra-fast position sensitive detectors for fast time-resolved measurements using the pulsed character of synchrotron radiation and novel X-ray laser sources.
References
Attwood D (1999) Soft X-rays and extreme ultraviolet radiation: principle and applications. Cambridge University Press. Cambridge, UK
Stöhr J (1992) NEXAFS spectroscopy. Springer Verlag, Berlin
Carr GL (2001) Rev Sci Instrum 72:1613
Müller RO (1972) Spectrochemical analysis by X-ray fluorescence. Plenum, New York
Siegbahn K (1990) J Electr Spectr Rel Phenom 51:11
Kirz J, Jacobsen C, Howells M (1995) Soft-x-ray microscopes and their biological applications. Q Rev Biophys 28:33–130
Niemann B, Rudolph D, Schmahl G (1974) Optics Comm 12:160–163
Bauer E (1994) Rep Prog Phys 57:895
Ade H (ed) (1997) J Electr Spectr Rel Phenom 84
Günther S, Kaulich B, Gregoratti L, Kiskinova M (2002) Prog Surf Sci 70:187–260
Spiller E (1995) Soft x-ray optics. SPIE, Bellingham
Howells M, Jacobsen C, Warwick T (2007) Principles and applications of zone plate microscopies. In: PW Hawkes, JCH John (eds) Science of microscopy. Springer Verlag, XXXVI
Schmahl G, Rudolph D, Schneider G, Guttmann P, Niemann B (1994) Phase contrast x-ray microscopy studies. Optik 97:181–182
Schneider G, Anderson E, Vogt S, Knochel C, Weiss D, Legros M, Larabell C (2002) Surf Rev Lett 9:177–183
Susini J, Salome M, Fayard B, Ortega R, Kaulich B (2002) Surf Rev Lett 9:203–211
Adams F, Van Vaeck L, Barrett R (2005) Spectrochim Acta B Atom Spectrosc 60:13–26
Braun A, Huggins FE, Shah N, Chen Y, Wirick S, Mun SB, Jacobsen C, Huffman GP (2005) Carbon 43:117
Denbeaux G, Anderson E, Chao W, Eimuller T, Johnson L, Kohler M, Larabell C, Legros M, Fischer P, Pearson A, Schultz G, Yager D, Attwood D (2001) Nucl Instrum Meth A 467:841–844
Kaulich B, Bacescu D, Susini J, David C, DiFabrizio E, Morrison GR, Charalambous P, Thieme J, Wilhein T, Kovac J, Cocco D, Salome M, Dhez O, Weitkamp T, Cabrini S, Cojoc D, Gianoncelli A, Vogt U, Podnar M, Zangrando M, Zacchigna M, Kiskinova M (2006) Proceedings of the 8th conference of X-ray microscopy, IPAP Series 7:22
Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M (2007) EMBO J 26:424–435
Thieme J et al, unpublished data
Gianoncelli A, Morrison GR, Kaulich B, Bacescu D, Kovac J (2006) Appl Phys Lett 89:251117
Palmer JR, Morrison GR (1991) OSA proceedings series, vol 11. Optical Society of America, Washington, DC, pp 141–145
Feser M, Jacobsen C, Rehak P, DeGeronimo G, Susini J, Joyeux D, Polack F (2004) Proceedings of the 7th international conference of X-ray microscopy, pp 529–534
Eichert D, Salome M, Bleuet P, Bohic S, Susini J (2006) IPAP Conf Series 7:210–212
Eichert D, Sfihi H, Combes C, Rey C (2004) Trans Tech Publications 256:927–931
Eichert D, Salomé M, Banu M, Susini J, Rey C (2005) Spectrochim Acta B 60:850–858
Chaboy J, Quartieri S (1995) Phys Rev B 52:6349–6357
Suzuki S, Watanabe Y, Ogino T, Heun S, Gregoratti L, Barinov A, Kaulich B, Kiskinova M, Zhu W, Bower C, Zhou O (2002) Phys Rev B 66:35414
Kovac J, Zalar A, Remskar M, Mrzel A, Mihailovic D, Gregoratti L, Kiskinova M (2003) Elettra Highlights
Barinov A, Gregoratti L, Kaulich B, Kiskinova M, Rizzi A (2001) Appl Phys Lett 79:2752–2754
Barinov A, Gregoratti L, Kiskinova M (2001) Phys Rev B 64:201312
Barinov A, Gregoratti L, Casalis L, Kiskinova M (2002) J Vac Sci Technol B 20:1918–1922
Melpignano P, Baron-Toaldo A, Biondo V, Priante S, Zamboni R, Murgia M, Caria S, Gregoratti L, Barinov A, Kiskinova M (2005) Appl Phys Lett 85:41105
Dumas P, Miller L (2003) Vibr Spectrosc 32:3–21
Carr GL, Chubar O, Dumas P (2006) In: Bhargava R, Levin IW (eds) Spectrochemical analysis using infrared detectors. Blackwell, pp 56–84
Moss D, Gashavora B, Mathis YL (2006) Infrared Phys Technol 49:53–56
Acknowledgements
We are grateful to O. Burrone and M. Campagna of ICGEB, Trieste, for providing cell samples for IR spectromicroscopy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eichert, D., Gregoratti, L., Kaulich, B. et al. Imaging with spectroscopic micro-analysis using synchrotron radiation. Anal Bioanal Chem 389, 1121–1132 (2007). https://doi.org/10.1007/s00216-007-1532-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-007-1532-4