
Abstract
Separation and detection of seven V-type (venomous) and G-type (German) organophosphorus nerve agent degradation products by gas chromatography with inductively coupled plasma mass spectrometry (GC–ICPMS) is described. The nonvolatile alkyl phosphonic acid degradation products of interest included ethyl methylphosphonic acid (EMPA, VX acid), isopropyl methylphosphonic acid (IMPA, GB acid), ethyl hydrogen dimethylamidophosphate sodium salt (EDPA, GA acid), isobutyl hydrogen methylphosphonate (IBMPA, RVX acid), as well as pinacolyl methylphosphonic acid (PMPA), methylphosphonic acid (MPA), and cyclohexyl methylphosphonic acid (CMPA, GF acid). N-(tert-Butyldimethylsilyl)-N-methyltrifluroacetamide with 1% TBDMSCl was utilized to form the volatile TBDMS derivatives of the nerve agent degradation products for separation by GC. Exact mass confirmation of the formation of six of the TBDMS derivatives was obtained by GC–time of flight mass spectrometry (TOF-MS). The method developed here allowed for the separation and detection of all seven TBDMS derivatives as well as phosphate in less than ten minutes. Detection limits for the developed method were less than 5 pg with retention times and peak area precisions of less than 0.01 and 6%, respectively. This method was successfully applied to river water and soil matrices. To date this is the first work describing the analysis of chemical warfare agent (CWA) degradation products by GC–ICPMS.

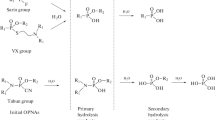
Illustrated here are six parent organophosphorus nerve agents corresponding to the degradation products analyzed by gas chromatography with ICPMS and ToF-MS detection. The authors would like to thank Daisy-Malloy Hamburg and Kevin M. Kubachka for creating this figure
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Chemical Weapons Convention (CWC), which came into effect on April 29, 1997, bans the production, acquisition, and direct or indirect transfer of chemical weapons, and also mandates the destruction of all chemical weapons held in reserve for member states [1]. The state members of the CWC created the Organization for the Prohibition of Chemical Weapons (OPCW) to help reach the objectives of the CWC. As of November 2006, the OPCW comprised 180 member states, six signatory states, and nine nonsignatory states, corresponding to 98% of the global population [2]. These toxic chemicals, which include schedule 1 blood, choking, blistering, and nerve agents, threaten not only the human population but also important food and water resources. According to the OPCW, the United States recently met a treaty deadline for the destruction of 50% of their chemical weapons held in reserve [3]. Due to the large worldwide reserves of such agents as well as the threat of terrorist attacks, the development of new analytical techniques for the analysis of chemical warfare agents (CWA) directly or by the trail left by their degradation metabolites is of vital importance for bolstering our capabilities to deal with CWA storage/destruction, as well as homeland security issues.
Organophosphorus nerve agent degradation products are nonvolatile, highly polar species with no good chromophore for UV detection (Fig. 1a and b). Due to the highly specific hydrolysis pathway of the parent CWA, analysis of the less toxic degradation products provides an alternative technique for CWA detection (Table 1). Previous studies have successfully utilized analytical methods such as gas chromatography/mass spectrometry (GC–MS) [4–10], gas chromatography/flame photometric detection (GC–FPD) [11], ion mobility/mass spectrometry (IMMS) [12, 13], capillary electrophoresis/ flame photometric detection (CE–FPD) [14, 15], 1-D 1H-31P inverse NMR spectroscopy [16, 17], and liquid chromatography/mass spectrometry (LC–MS) [18] for the analysis of CWA agent degradation products with detection limits in the ng mL−1 range. Chemical warfare degradation product analysis by inductively coupled plasma mass spectrometry (ICPMS) coupled with ion-pairing reversed phase high-performance liquid chromatography (IP–RP–HPLC) has recently been shown to be a rapid and reliable speciation technique with detection limits in the ng L−1 range [19].


Hydrolysis pathways of G- (a) and V- (b) type nerve agents
A major advantage of element-specific detection with ICPMS is the ability to couple this detector with multiple separation techniques, including liquid chromatography, capillary electrophoresis, and gas chromatography [19–22]. Recently, gas chromatography with ICPMS detection has proven to be a powerful technique for phosphorus speciation studies [20–22]. Shah et al. recently investigated the use of solid-phase microextraction with GC–ICPMS for the analysis of phosphoric acid triesters in human plasma with detection limits in the ng L−1 range [23]. The use of gas chromatography for the separation of chemical warfare agent degradation products has been well documented for more than fifteen years [5, 7, 9, 24–30]. Due to the nonvolatile nature of these moderate to highly polar phosphonic acid degradation products, multiple types of derivatization techniques, including pentafluorobenzyl esters, methyl esters, trimethylsilyl, and tert-butyldimethylsilyl esters, have been studied for the generation of more volatile species amenable for GC separation [28]. Black et al. reviewed derivatization reactions for the analysis of chemical warfare agents and their degradation products [28]. This review described both trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBDMS) derivatives as being the most popular for phosphonic acids, with the TBDMS derivatives suspected to be the most stable and least sensitive to moisture [28]. Typically, detection of these CWA degradation product derivatives separated by GC has been accomplished with FPD, NPD, AED, quad-MS, and ion trap MS [28]. To date, the use of GC–ICPMS for the analysis of CWA degradation product derivatives has never been reported in the literature.
In this work, the application of GC–ICPMS to the analysis of common degradation products of the chemical warfare agents Soman, Sarin, Tabun, RVX and VX is described. Optimization of temperature and time for the formation of TBDMS derivatives of seven degradation products is described. Analytical figures of merit for each species studied (ethyl methylphosphonic acid, isopropyl methylphosphonic acid, methylphosphonic acid, cyclohexyl methylphosphonic acid, ethyl hydrogen dimethylamidophosphate sodium salt, isobutyl hydrogen methylphosphonate, and pinacolyl methylphosphonic acid) are presented. Finally, the method developed here was applied to river water and soil matrices to determine the sensitivity of ICPMS for these organophosphorus nerve agent degradation product derivatives.
Experimental
Reagents
Seven chemical warfare degradation products were obtained from Cerilliant (Austin, TX, USA) as 1 mg mL−1 certified reference materials (CRMs). CRMs are used as standard analytical solutions for the analysis of Schedule 1, 2, or 3 toxic chemicals, their precursors, and/or degradation products as mandated by the CWC for verification [1]. The degradation products utilized for these experiments included ethyl methylphosphonic acid (EMPA, VX acid), isopropyl methylphosphonic acid (IMPA, GB acid), methylphosphonic acid (MPA), cyclohexyl methylphosphonic acid (CMPA, GF acid), ethyl hydrogen dimethylamidophosphate sodium salt (EDPA, GA acid), isobutyl hydrogen methylphosphonate (IBMPA, RVX acid), and pinacolyl methylphosphonic acid (PMPA). N-(tert-Butyldimethylsilyl)-N-methyltrifluroacetamide with 1% TBDMSCl (Sigma-Aldrich, St. Louis, MO, USA) was utilized as the derivatizing reagent. Derivatization reactions were performed with distilled acetonitrile (Tedia, Cincinnati, OH, USA).
Helium gas (Matheson Gas Products, Parisppany, NJ, USA) with a purity of 99.999% with 1% Xe was used as the GC carrier gas throughout all experiments. Xenon served as the tuning gas for optimizing the GC–ICPMS parameters prior to analysis.
Derivatization
Due to the nonvolatile nature of these CWA degradation products, derivatization was required in order to achieve volatile species for separation with gas chromatography. TBDMS derivatives were formed by creating a 2:1 mixture of acetonitrile with derivatizing agent. Figure 2 describes the esterification reaction for the formation of the TBDMS derivatives. Stock solutions consisting of a 40 μg mL−1 mixture of each CWA degradation product was created through dilution of the stock degradation products into the acetonitrile/derivatizing agent mixture. Optimization of the derivatization time was based upon the peak area of duplicate injections of a 40 μg mL−1 mixture heated at 60 °C for 15, 30, 45, and 60 min, with 45 min chosen as the optimum (Fig. 3a). Temperature optimization was then preformed at room temperature, 40, 60, and 80 °C for 45 minutes with 80 °C chosen as the optimum temperature for the derivatization (Fig. 3b).

TBDMS esterification of alkyl phosphonic acids

a and b -Optimization of derivatization time and temperature
Environmental samples
Investigation into the tolerance of the method for complex environmental sample matrices led to the collection of river water and soil samples. In a typical real world setting this method would only be required to monitor a few of the degradation products in order to confirm proper storage and disposal. The development of a more versatile method capable of separating degradation products from multiple CWA provides future researchers with the flexibility to adjust the method as desired.
River water samples were collected in polypropylene bottles from the Little Miami River (Cincinnati, OH, USA). Initial experiments consisted of drying a 1 mL aliquot of river water under nitrogen. The dried river water was reconstituted in 250 μL of acetonitrile, vortexed, and spiked with a derivative mixture of degradation products to yield a concentration of 20 μg mL−1. The resulting solution was filtered with 0.20 μm nylon–nitrocelluose acetate (CA) filters prior to analysis. Additional experiments consisted of taking 100 μL of river water and spiking with 50 μL aliquots of each 1000 μg mL−1 stock degradation product. The resulting solution was dried and reconstituted with 600 μL acetonitrile and 300 μL derivatizing agent to yield a 55.5 μg mL−1 mixture. The mixture was heated at 80 °C for 45 min followed by filtering prior to analysis.
Soil samples were collected from outside the laboratory at the University of Cincinnati. Initial experiments consisted of taking 1 g soil and adding 1 mL of distilled deionized water.The resulting slurry was stirred and then the water extract was removed and dried under nitrogen. The dried soil extract was reconstituted in 250 uL of acetonitrile, vortexed and spiked with a derivative mixture of degradation products to yield a concentration of 20 μg mL−1. The mixture was then filtered with 0.20 um nylon cellulose syringe filters prior to analysis. Additional experiment consisted of taking a 100 μL of soil extract and spiking with 50 μL aliquots of each 1000 μg mL−1 stock degradation product. The spiked solution was dried and then reconstituted with 600 μL acetonitrile and 300 μL derivatizing agent to yield a 55.5 μg mL−1 mixture. The mixture was heated at 80 °C for 45 min followed by filtering prior to analysis.
Instrumentation
Gas chromatography (GC)
GC with ICP–MS detection is a highly desirable mode of operation, since the gaseous argon plasma detector works best with gaseous sample introduction. While liquid sample introduction is the most widely used, the plasma must desolvate the aerosol prior to analyte ionization, requiring time and energy that might have gone into forming analyte ions. With gas sample introduction, 10× lower detection levels are routinely obtained over liquid sample introduction. An Agilent 6890 (Agilent Technologies, Palo Alto, CA, USA) gas chromatograph with helium carrier gas with 1% xenon was used to separate the seven chemical warfare degradation products. Xenon gas allowed for daily instrument parameter optimization prior to analysis. An HP-5 (5%-phenyl-methyl-polylsiloxane) capillary column (30 m, 0.32 mm i.d., 0.25 μm film thickness) was used for all separation experiments. A detailed description of the GC parameters is provided in Table 2.
Inductively coupled plasma mass spectrometer (ICPMS)
An Agilent 7500ce (Agilent Technologies, Tokyo, Japan) ICPMS equipped with shield torch and collision/reaction cell technology was used for the element-specific detection of 31P and 47 PO+ throughout method development. The reason for monitoring 47PO+ is to ensure that no 31P signal was lost due to oxide formation; however, significant oxide formation was not observed at any time during this experiment. Due to the gaseous sample introduction technique that accompanies GC–ICPMS, the atmospheric polyatomic interferences (14N16O1H+ and 15N16O+) that are common observed with LC–ICPMS for 31P are not as abundant, resulting in a lower background signal at m/z = 31. Due to this lower background signal, the collision/reaction cell was not needed to reduce the m/z = 31 background in these experiments. Electronic coupling of the ICPMS with the GC was accomplished through the use of a remote cable, which allowed data acquisition to be started simultaneously prior to each separation experiment. Instrument parameters were optimized daily prior to analysis by optimizing the Xe signal at m/z = 124. A detailed description of the ICPMS parameters used is provided in Table 2.
Gas chromatography–time of flight mass spectrometry (GC–TOF-MS)
An Agilent 6890 (Agilent Technologies, Palo Alto, CA, USA) gas chromatograph equipped with a Micromass GCT (Waters Corporation, Milford, MA, USA) time of flight mass spectrometer (TOF-MS) was utilized for exact mass confirmation of the TBDMS derivatives. A J&W Scientific (Agilent Technologies, Palo Alto, CA, USA) capillary column (D-XLB; 30 m, 0.25 mm i.d., 0.25 μm film thickness) was utilized for all separation experiments. Detector parameters consisted of a source temperature of 180 °C, an electron energy of 70 eV, a trap current of 150 μA, and an MCP voltage of 2350 V. Tuning of the TOF-MS was accomplished through the use of perfluorotributylamine (FC-43; Scientific Instrument Services, Ringoes, NJ, USA).
Results and discussion
Elemental speciation analysis by ICPMS is a powerful analytical technique due to the high sensitivity and selectivity offered by this instrument. Phosphorus analysis by ICPMS provides an ionization source capable of overcoming the high first ionization potential (10.5 eV) of this element as well as state-of-the-art ion optics and an octopole interference reduction system.
Previous analysis of organophosphorus CWA degradation products by LC–ICPMS provided improved sensitivity, but a lack of chromatographic resolution resulted in interferences from phosphate in environmental sample matrices [19]. Efforts to improve the chromatographic resolution have led to an investigation of gas chromatography followed by 31P element-specific detection with ICPMS. Due to the nonvolatile nature of the organophosphorus CWA degradation products, formation of the volatile TBDMS ester was required. TBDMS was chosen as the derivatization method of choice due to its stability and tolerance to trace amounts of moisture [28]. Due to the selectivity of ICPMS for 31P, an efficient derivatization reaction was vital in order to prevent intermediate species formation. Optimum reaction conditions for the formation of the TBDMS derivatives of alkyl phosphonic CWA degradation products were determined to be 80 °C for 45 min (Fig. 3a and b). These conditions allowed for the separation of EMPA (VX acid), IMPA (GB acid), EDPA (GA acid), IBMPA (RVX acid), PMPA, MPA, and CMPA (GF acid) in less than nine minutes. Figure 4 shows the separation and detection of a 5-ng mixture of the seven CWA degradation products. To date, this is the first work utilizing GC–ICPMS for the analysis of TBDMS derivatives of organophosphorus CWA.

Separation of a 5 ng mixture of seven TBDMS derivatives
GC–TOF-MS
Confirmation of the formation of TBDMS derivatives for six of the CWA degradation products of interest was determined by GC–TOF-MS, as described in the “Experimental” section. Electron impact (EI) ionization of silyl derivatives (TBDMS and TMS) of alkyl phosphonic acids in the positive mode typically results in the formation of a base peak at m/z 153 corresponding to [M–C n H2n –Me]+ and [M–C n H2n–Bu]+ [28]. Loss of [M–CH3]+ and [M–C4H9]+ also provides higher mass ions with weak to moderate intensities [28]. Figure 5a–f shows the exact mass confirmation for the formation of TBDMS derivatives of EMPA (VX acid), IMPA (GB acid), IBMPA (RVX acid), PMPA, MPA, and CMPA (GF acid); the masses observed correlate well to their exact calculated masses.



a–f Exact mass confirmations of TBDMS derivatives by GC–TOF-MS
The TBDMS derivative of ethyl hydrogen dimethylamidophosphate sodium salt (EDPA, GA acid) posed difficulties for exact mass confirmation by GC–TOF-MS. EDPA possesses both highly acidic and highly basic pK a values, as described in Table 1. Retention time of the individual TBDMS derivative was confirmed by GC–ICPMS, but interpretation of the mass spectrum was not achieved due to complex gas phase chemistry and rearrangement. Efforts to confirm the formation of this TBDMS derivative are currently underway.
Analytical figures of merit
Calibration curves ranging from 0.156 to 20 ng (nerve agent derivative injected on-column) were prepared through serial dilution of standard mixtures of EMPA, IMPA, EDPA, IBMPA, PMPA, MPA, and CMPA TBDMS derivatives. Correlation coefficients (R 2) ranged from 0.998–0.999 and demonstrated excellent linearity for each species analyzed. Detection limits (3σ) calculated based upon three times the standard deviation of seven replicates of the blank peak area (IUPAC) ranged from 1.72–4.92 pg for all seven TBDMS derivatives. The precision, based upon seven replicate injections of a 5 ng mixture of TBDMS derivatives on-column, was less than 0.01% for retention time and less than 5.72% for peak area. A summary of the analytical figures of merit is provided in Table 3. Table 4 provides a detection limit comparison of LC–ICPMS to the GC–ICPMS method developed here [19]. The improvement in detection limits for the GC–ICPMS method are attributed to the lower background associated with this gaseous sample introduction technique compared to conventional liquid introduction (Table 4).
Phosphate and environmental samples
Application of the method developed here to complex sample matrices consisted of preliminary experiments to determine the derivatization, separation, and detection of phosphate–TBDMS in a mixture also containing the seven degradation products. Figure 6 shows the separation of a 30 ng on-column mixture of the seven degradation products and phosphate–TBDMS in less than ten minutes. The ability to derivatize, separate, and detect inorganic phosphate permitted the application of the developed method to river water and soil matrices.

Standard separation of 30 ng mixture containing phosphate–TBDMS
Application of the developed method to river water consisted of the preparation of two separate sample types. First, a standard derivative mixture was spiked into dried river water sample to determine the sensitivity of the developed method to a complex matrix. Figure 7a demonstrates the separation and detection of all seven degradation products spiked into the river water matrix. Following the confirmation of minimal matrix effects upon derivative response, derivatization within the complex matrix was explored. River water extracts were prepared, spiked and derivatized following the procedure described in the “Experimental” section. Figure 7b illustrates the separation and detection of the seven derivatized degradation products as well as phosphate from the river water in less than ten minutes.

Detection of spiked degradation products (a) and spiked derivatized degradation products (b) in river water samples
Application of the developed method to soil samples followed the same procedure as that used for the river water samples. Two types of soil extract samples were prepared. First, the sensitivity of the developed method to the complex soil matrix was investigated by spiking the soil extract with a standard derivatized mixture, as described in the “Experimental” section. Figure 8a shows the separation of a 20 ng on-column mixture of the seven degradation products. The confirmation of minimal matrix effects in the spiked sample led to an analysis of the effects of the matrix on the derivatization reaction. Soil extracts were spiked and derivatized following the procedure described in the “Experimental” section. Figure 8b shows the separation and detection of all seven derivatized degradation products and phosphate in less than ten minutes.

Detection of spiked degradation products (a) and spiked derivatized degradation products (b) in soil samples
Conclusion
In this work, gas chromatography coupled with ICPMS allowed for the separation and detection of seven organophosphorus nerve agent degradation products and phosphate in less than ten minutes. Through a simple esterification reaction, the nonvolatile degradation products were converted into their TBDMS derivatives, which were ideal for GC separation. The method developed here provides a highly sensitive and selective technique yielding detection limits of less than 5 pg and retention time and peak area precisions of less than 0.01 and 6%, respectively. The method was successfully applied to complex river water and soil matrices. Future studies currently underway include the investigation of alternative derivatization techniques, including those utilizing TMS, p-bromophenol and pentafluorbenzyl esters, as well as the use of solid-phase microextraction (SPME) preconcentration to analyze these CWA degradation products.
References
OPCW (1993) Convention on the prohibition of the development, production, stockpiling and use of chemical weapons and on their destruction. Organization for the Prohibition of Chemical Weapons (OPCW), The Hague, The Netherlands
OPCW (2006) Weapons. Accessed September 27, 2006. http://www.opcw.org/docs/cwc_eng.pdf
Ember LR (2006) ACS Chem Eng News 84:87–89
Tornes JA, Opstad AM, Johnsen BA (2006) Sci Total Environ 356:235–246
Barr JR, Driskell WJ, Aston LS, Martinez RA (2004) J Anal Toxicol 28:372–378
Smith JR, Schlager JJ (1996) J High Res Chromatogr 19:151–154
Vasilevskii SV, Kireev AF, Rybal’chenko IV, Suvorkin VN (2002) J Anal Chem USSR 57:491–497
Saradhi UVRV, Prabhakar S, Reddy TJ, Vairamani M (2006) J Chromatogr A 1129:9–13
Gupta AK, Pardasani D, Kanaujia PK, Tak V, Dubey DK (2006) Rapid Commun Mass Spectrom 20:2115–2119
Lee HSN, Basheer C, Lee HK (2006) J Chromatogr A 1124:91–96
Logan TP, Allen ED, Way MR, Swift AT, Soni S-D, Koplovitz I (2006) Toxicol Mech Methods 16:359–363
Steiner WE, Clowers BH, Matz LM, Siems WF, Hill HH Jr (2002) Anal Chem 74:4343–4352
Steiner WE, Harden CS, Hong F, Klopsch SJ, Hill HH, McHughVM (2006) J Am Soc Mass Spectrom 17:241–245
Hooijschuur EWJ, Kientz CE, Brinkman UAT (2001) J Chromatogr A 928:187–199
Pumera M (2006) J Chromatogr A 1113:5–13
Meier UC (2004) Anal Chem 76:392–398
Koskela H, Grigoriu N, Vanninen P (2006) Anal Chem 78:3715–3722
Wada T, Nagasawa E, Hanaoka S (2006) Appl Organomet Chem 20:573–579
Richardson DD, Sadi BBM, Caruso JA (2006) J Anal Atom Spectrom 21:396–403
Shah M, Meija J, Cabovska B, Caruso JA (2006) J Chromatogr A 1103:329–336
Vonderheide AP, Meija J, Montes-Bayon M, Caruso JA (2003) J Anal Atom Spectrom 18:1097–1102
Proefrock D, Leonhard P, Wilbur S, Prange A (2004) J Anal Atom Spectrom 19:623–631
Shah M, Caruso JA (2005) J Sep Sci 28:1969–1984
Purdon JG, Pagotto JG, Miller RK (1989) J Chromatogr 475:261–272
Black RM, Clarke RJ, Read RW, Reid MTJ (1994) J Chromatogr A 662:301–321
Creasy WR, Rodriguez AA, Stuff JR, Warren RW (1995) J Chromatogr A 709:333–344
Sng MT, Ng WF (1999) J Chromatogr A 832:173–182
Black RM, Muir B (2003) J Chromatogr A 1000:253–281
Crenshaw MD, Cummings DB (2004) Phosphorus Sulfur 179:1009–1018
Wang Q, Xie J, Gu M, Feng J, Ruan J (2005) Chromatographia 62:167–173
Acknowledgements
The authors would like to thank Agilent technologies for their continued support of our research, as well as NIEHS Grant ES04908 (supplement 3P42-E5004908-15S4).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Richardson, D.D., Caruso, J.A. Derivatization of organophosphorus nerve agent degradation products for gas chromatography with ICPMS and TOF-MS detection. Anal Bioanal Chem 388, 809–823 (2007). https://doi.org/10.1007/s00216-007-1164-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-007-1164-8